Creating Long-Format Peak Tables with Optional Annotations
Source:vignettes/long-format-peaklist.Rmd
long-format-peaklist.RmdIntroduction
The tidyXCMS package provides functions to work with
XCMS metabolomics data in a tidy, long-format structure. This vignette
demonstrates how to use the tidy_peaklist() function to
create peak tables from XCMS results.
The resulting long-format table has one row per feature per sample, integrating:
- Peak detection results from XCMS
- Feature grouping (correspondence) information
- Sample metadata
This structure makes the data ideal for downstream analysis with
tidyverse tools like dplyr and ggplot2.
You can optionally enhance these tables with additional annotations, which we’ll explore later in this vignette.
Setup
library(tidyXCMS)
library(xcms)
library(BiocParallel)
library(dplyr)
library(ggplot2)
library(tidyr)
library(MsExperiment)
# Optional packages for enhanced functionality
# Install if needed: BiocManager::install("CAMERA") or install.packages("commonMZ")
if (requireNamespace("MSnbase", quietly = TRUE)) library(MSnbase)
if (requireNamespace("CAMERA", quietly = TRUE)) library(CAMERA)
if (requireNamespace("MsFeatures", quietly = TRUE)) library(MsFeatures)
if (requireNamespace("commonMZ", quietly = TRUE)) library(commonMZ)
if (requireNamespace("ggalluvial", quietly = TRUE)) library(ggalluvial)
if (requireNamespace("RColorBrewer", quietly = TRUE)) library(RColorBrewer)
if (requireNamespace("ggpie", quietly = TRUE)) library(ggpie)Example Workflow
Load Example Data
We’ll use the xmse dataset from XCMS, which contains
preprocessed LC-MS data from wild-type and knockout samples. This
dataset already has peak detection and feature grouping completed.
# Load example data from XCMS (already preprocessed)
xdata <- loadXcmsData("xmse")Examine Sample Metadata
Let’s check what sample information is available:
sampleData(xdata)
#> DataFrame with 8 rows and 4 columns
#> sample_name sample_group spectraOrigin sample_type
#> <character> <character> <character> <character>
#> 1 ko15 KO /usr/local... QC
#> 2 ko16 KO /usr/local... study
#> 3 ko21 KO /usr/local... study
#> 4 ko22 KO /usr/local... QC
#> 5 wt15 WT /usr/local... study
#> 6 wt16 WT /usr/local... study
#> 7 wt21 WT /usr/local... QC
#> 8 wt22 WT /usr/local... study
# Check results
cat("Dataset contains", nrow(chromPeaks(xdata)), "peaks across",
length(unique(chromPeaks(xdata)[, "sample"])), "samples,",
"grouped into", nrow(featureDefinitions(xdata)), "features\n")
#> Dataset contains 3651 peaks across 8 samples, grouped into 351 featuresFix File Paths
The example data contains file paths from the original system. We need to update them to match the current system:
# Get the correct base path for faahKO package on this system
cdf_path <- file.path(find.package("faahKO"), "cdf")
# Find all CDF files recursively in the cdf_path directory
real_paths <- list.files(cdf_path, recursive = TRUE, full.names = TRUE)
# Create a mapping table using basenames for safe matching
path_mapping <- tibble(
real_path = real_paths,
basename_file = basename(real_paths)
)
# Join with spectra dataOrigin by basename and replace
spectra_df <- tibble(dataOrigin = spectra(xdata)$dataOrigin) %>%
mutate(basename_file = basename(dataOrigin)) %>%
left_join(path_mapping, by = "basename_file")
spectra(xdata)$dataOrigin <- spectra_df$real_pathCreating a Basic Long-Format Peak Table
Let’s start by creating a long-format peak table directly from the XCMS results. This gives us all the essential peak information in a tidy format.
# Create basic peak table
peak_table <- tidy_peaklist(xdata)
# Check the structure
dim(peak_table)
#> [1] 2808 28
# View first few rows
head(peak_table)
#> # A tibble: 6 × 28
#> sample_name sample_group spectraOrigin sample_type fromFile feature_id f_mzmed
#> <chr> <chr> <chr> <chr> <dbl> <int> <dbl>
#> 1 ko15 KO /usr/local/l… QC 1 1 200.
#> 2 ko15 KO /usr/local/l… QC 1 2 205
#> 3 ko15 KO /usr/local/l… QC 1 3 206
#> 4 ko15 KO /usr/local/l… QC 1 4 207.
#> 5 ko15 KO /usr/local/l… QC 1 5 233
#> 6 ko15 KO /usr/local/l… QC 1 6 241.
#> # ℹ 21 more variables: f_mzmin <dbl>, f_mzmax <dbl>, f_rtmed <dbl>,
#> # f_rtmin <dbl>, f_rtmax <dbl>, ms_level <int>, filepath <chr>,
#> # filename <chr>, peakidx <dbl>, mz <dbl>, mzmin <dbl>, mzmax <dbl>,
#> # rt <dbl>, rtmin <dbl>, rtmax <dbl>, into <dbl>, intb <dbl>, maxo <dbl>,
#> # sn <dbl>, is_filled <lgl>, merged <lgl>
# Check column names
colnames(peak_table)
#> [1] "sample_name" "sample_group" "spectraOrigin" "sample_type"
#> [5] "fromFile" "feature_id" "f_mzmed" "f_mzmin"
#> [9] "f_mzmax" "f_rtmed" "f_rtmin" "f_rtmax"
#> [13] "ms_level" "filepath" "filename" "peakidx"
#> [17] "mz" "mzmin" "mzmax" "rt"
#> [21] "rtmin" "rtmax" "into" "intb"
#> [25] "maxo" "sn" "is_filled" "merged"The resulting table contains all feature and peak information from XCMS, organized with one row per feature per sample.
Enhancing Peak Tables with CAMERA Annotations
CAMERA can annotate isotopes, adducts, and group features into pseudospectra, providing valuable chemical context to your peak table.
Note: CAMERA requires an xcmsSet
object. Since our data is an XcmsExperiment, we first
convert it to XCMSnExp and then to
xcmsSet.
Creating a CAMERA object
In this first step we create a CAMERA object from the XCMS object. We specify the polarity to help CAMERA determine appropriate adducts and fragments.
# Convert XcmsExperiment to xcmsSet for CAMERA (required for compatibility)
# Two-step conversion: XcmsExperiment -> XCMSnExp -> xcmsSet
xset <- xdata %>%
as("XCMSnExp") %>%
as("xcmsSet")
# Create xsAnnotate object with polarity
xs <- xsAnnotate(xset, polarity = "positive")Grouping coeluting peaks
The first step to grouping features is to group co-eluting peaks. This is a naïve approach that we will refine later.
# Group peaks by retention time
xs <- groupFWHM(xs, perfwhm = 0.1, intval = "into", sigma = 6)
#> Start grouping after retention time.
#> Created 226 pseudospectra.Grouping based on correlation
Now we group the features based on correlations. This looks at each group from the previous step and splits them into separate groups for peaks that correlate with each other.
Note: For this small example dataset, don’t use correlation across samples as it is unreliable with this few samples.
# Group by correlation
xs <- groupCorr(xs, calcIso = FALSE, calcCiS = TRUE, calcCaS = FALSE, cor_eic_th = 0.7, pval = 1E-6)
#> Start grouping after correlation.
#> Generating EIC's ..
#>
#> Calculating peak correlations in 226 Groups...
#> % finished: 10 20 30 40 50 60 70 80 90 100
#>
#> Calculating graph cross linking in 226 Groups...
#> % finished: 10 20 30 40 50 60 70 80 90 100
#> New number of ps-groups: 271
#> xsAnnotate has now 271 groups, instead of 226Isotope annotation
This annotates peaks that are possible isotopes based on m/z difference and intensity patterns.
# Find isotopes
xs <- findIsotopes(xs, ppm = 10, mzabs = 0.01, intval = "into", maxcharge = 2)
#> Generating peak matrix!
#> Run isotope peak annotation
#> % finished: 10 20 30 40 50 60 70 80 90 100
#> Found isotopes: 29Annotation of adducts and fragments
Now we try to annotate adducts and fragments based on expected mass
differences. We’ll use the commonMZ package to generate a
comprehensive list of adduct and fragment rules for CAMERA.
First, we get the adduct/fragment rules for positive mode:
# warn_clash=TRUE will warn about indistinguishable annotations (e.g., [M+NH4]+ vs -NH3)
# Remove [M+NH4]+ if it's less common in your data
rules_pos <- MZ_CAMERA(mode = "pos", warn_clash = TRUE, clash_ppm = 5) %>%
filter(name != "[M+NH4]+") %>%
as.data.frame()
#> Warning in MZ_CAMERA(mode = "pos", warn_clash = TRUE, clash_ppm = 5): The following adducts/fragments seem to collide.
#> # A tibble: 2 × 2
#> first second
#> <chr> <chr>
#> 1 [M+H-NH3]+ [M+NH4]+
#> 2 [M+H-C3H4]+ [M+H+(CH3)2CO-H2O]+ (acetone cond.)
#>
#>
#> Consider removing one of them. Example:
#> rules=rules[ !grepl("[M+NH4]+",rules[,"name"],fixed=TRUE) ,]Now we can annotate adducts using these rules:
# Find adducts using the rules from commonMZ
xs <- findAdducts(xs, ppm = 500, mzabs = 0.2, multiplier = 4, polarity = "positive", rules = rules_pos)
#> Generating peak matrix for peak annotation!
#> Polarity is set in xsAnnotate: positive
#> Found and use user-defined ruleset!
#> Calculating possible adducts in 271 Groups...
#> % finished: 10 20 30 40 50 60 70 80 90 100Creating the Peak Table with CAMERA Annotations
Now we can create a peak table that includes all the CAMERA
annotations. We pass both the original xdata object and the
CAMERA annotations from xs:
peak_table_camera <- tidy_peaklist(xdata, xs)
# Check the structure
dim(peak_table_camera)
#> [1] 2808 31
colnames(peak_table_camera)
#> [1] "sample_name" "sample_group" "spectraOrigin" "sample_type"
#> [5] "fromFile" "feature_id" "f_mzmed" "f_mzmin"
#> [9] "f_mzmax" "f_rtmed" "f_rtmin" "f_rtmax"
#> [13] "ms_level" "isotopes" "adduct" "pcgroup"
#> [17] "filepath" "filename" "peakidx" "mz"
#> [21] "mzmin" "mzmax" "rt" "rtmin"
#> [25] "rtmax" "into" "intb" "maxo"
#> [29] "sn" "is_filled" "merged"The table now includes additional columns for CAMERA annotations:
isotopes, adduct, and pcgroup
(pseudospectrum correlation group).
Enhancing Peak Tables with MsFeatures Grouping
The MsFeatures package offers another way to enhance
peak tables by grouping features based on retention time similarity,
abundance correlation, or EIC similarity.
Why Use MsFeatures?
- Retention Time Grouping: Group co-eluting features that likely come from the same compound
- Abundance Correlation: Group features with correlated intensities across samples
- EIC Similarity: Group features based on similar extracted ion chromatograms
- Stepwise Refinement: Apply multiple grouping algorithms sequentially to refine groups
Applying Feature Grouping
The groupFeatures() function adds a
feature_group column to the feature definitions. Let’s
demonstrate with retention time-based grouping:
# Group features with similar retention times (within 10 seconds)
xdata_grouped <- groupFeatures(xdata, param = SimilarRtimeParam(diffRt = 10))
# Check feature groups in featureDefinitions
head(featureDefinitions(xdata_grouped)$feature_group)
#> [1] "FG.083" "FG.005" "FG.005" "FG.078" "FG.016" "FG.073"
# Count how many feature groups were created
table(featureDefinitions(xdata_grouped)$feature_group)
#>
#> FG.001 FG.002 FG.003 FG.004 FG.005 FG.006 FG.007 FG.008 FG.009 FG.010 FG.011
#> 4 6 2 4 6 5 2 4 3 4 3
#> FG.012 FG.013 FG.014 FG.015 FG.016 FG.017 FG.018 FG.019 FG.020 FG.021 FG.022
#> 6 4 2 3 3 3 4 3 2 5 2
#> FG.023 FG.024 FG.025 FG.026 FG.027 FG.028 FG.029 FG.030 FG.031 FG.032 FG.033
#> 2 4 5 2 5 2 3 4 2 2 4
#> FG.034 FG.035 FG.036 FG.037 FG.038 FG.039 FG.040 FG.041 FG.042 FG.043 FG.044
#> 2 2 6 3 2 2 4 2 2 4 4
#> FG.045 FG.046 FG.047 FG.048 FG.049 FG.050 FG.051 FG.052 FG.053 FG.054 FG.055
#> 2 3 2 3 3 5 2 3 4 2 6
#> FG.056 FG.057 FG.058 FG.059 FG.060 FG.061 FG.062 FG.063 FG.064 FG.065 FG.066
#> 2 6 4 2 4 3 3 2 2 2 2
#> FG.067 FG.068 FG.069 FG.070 FG.071 FG.072 FG.073 FG.074 FG.075 FG.076 FG.077
#> 3 2 2 3 2 3 2 3 3 3 3
#> FG.078 FG.079 FG.080 FG.081 FG.082 FG.083 FG.084 FG.085 FG.086 FG.087 FG.088
#> 2 3 3 3 2 3 3 3 3 3 2
#> FG.089 FG.090 FG.091 FG.092 FG.093 FG.094 FG.095 FG.096 FG.097 FG.098 FG.099
#> 3 2 2 2 2 3 2 4 2 3 3
#> FG.100 FG.101 FG.102 FG.103 FG.104 FG.105 FG.106 FG.107 FG.108 FG.109 FG.110
#> 3 3 2 2 2 2 2 2 2 2 2
#> FG.111 FG.112 FG.113 FG.114 FG.115 FG.116 FG.117 FG.118 FG.119 FG.120 FG.121
#> 2 2 1 1 1 1 1 1 1 1 1
#> FG.122 FG.123 FG.124 FG.125 FG.126 FG.127 FG.128 FG.129 FG.130 FG.131 FG.132
#> 1 1 1 1 1 1 1 1 1 1 1
#> FG.133
#> 1Creating Peak Table with MsFeatures Grouping
When you create a long-format peak table from an object with feature
groups, the feature_group column is automatically
included:
# Create peak table with feature grouping
peak_table_grouped <- tidy_peaklist(xdata_grouped)
# Check that feature_group column is present
"feature_group" %in% colnames(peak_table_grouped)
#> [1] TRUE
# View some feature groups
peak_table_grouped %>%
select(feature_id, feature_group, f_mzmed, f_rtmed) %>%
distinct() %>%
head(10)
#> # A tibble: 10 × 4
#> feature_id feature_group f_mzmed f_rtmed
#> <int> <chr> <dbl> <dbl>
#> 1 1 FG.083 200. 2903.
#> 2 2 FG.005 205 2790.
#> 3 3 FG.005 206 2789.
#> 4 4 FG.078 207. 2719.
#> 5 5 FG.016 233 3024.
#> 6 6 FG.073 241. 3683.
#> 7 7 FG.024 242. 3664.
#> 8 8 FG.113 244. 2833.
#> 9 9 FG.071 249. 3676.
#> 10 10 FG.072 250. 3675.Combining CAMERA and MsFeatures Annotations
You can also combine both approaches. Apply
groupFeatures() first, then proceed with CAMERA
annotation:
# Apply feature grouping
xdata_grouped <- groupFeatures(xdata, param = SimilarRtimeParam(diffRt = 10))
# Create peak table with both CAMERA and MsFeatures annotations
# Reuse the xs object created earlier (CAMERA annotations are the same)
peak_table_combined <- tidy_peaklist(xdata_grouped, xs)
# View combined annotations
peak_table_combined %>%
filter(adduct != "") %>%
select(feature_id, feature_group, f_mzmed, f_rtmed, isotopes, adduct, pcgroup) %>%
distinct(feature_id, .keep_all = TRUE) %>%
head()
#> # A tibble: 6 × 7
#> feature_id feature_group f_mzmed f_rtmed isotopes adduct pcgroup
#> <int> <chr> <dbl> <dbl> <chr> <chr> <int>
#> 1 3 FG.005 206 2789. "" [M+2K]2+ 334.047 [… 11
#> 2 6 FG.073 241. 3683. "" [M+H-S]+ 272.112 [… 87
#> 3 11 FG.073 255. 3682. "" [M+H-H2O]+ 272.112 87
#> 4 13 FG.001 266. 3669. "" [M+H-C2H4-CO2]+ 33… 44
#> 5 19 FG.005 279 2788. "" [M+H-C3H4O]+ 334.0… 11
#> 6 21 FG.001 281. 3669. "[1][M]+" [M+H-NH3-C3H4]+ 33… 44Now you have both:
-
feature_group: Groups features based on retention time and correlation across samples (from MsFeatures) -
pcgroup: Groups features into pseudospectra based on EIC similarity (from CAMERA) -
isotopesandadduct: Isotope and adduct annotations (from CAMERA)
This combined approach gives you multiple perspectives on which features might originate from the same compound.
We can also visually compare the mapping.
group_mapping <- peak_table_combined %>%
filter(adduct != "") %>%
select(feature_id, feature_group, f_mzmed, f_rtmed, isotopes, adduct, pcgroup) %>%
distinct(feature_id, .keep_all = TRUE)
# Determine ordering by the most common pairs
order_group_mapping <- group_mapping %>%
count(feature_group, pcgroup, sort = TRUE)
# Choose ordering to align best-matching pairs
left_order <- unique(order_group_mapping$feature_group)
right_order <- unique(order_group_mapping$pcgroup)
group_mapping <- group_mapping %>%
mutate(
feature_group = factor(feature_group, levels = left_order),
pcgroup = factor(pcgroup, levels = right_order)
)
ggplot(group_mapping, aes(axis1 = feature_group, axis2 = pcgroup, fill = feature_group)) +
geom_alluvium(alpha = 0.7) +
geom_stratum() +
geom_text(stat = "stratum", aes(label = after_stat(stratum))) +
theme_minimal() +
labs(x = "", y = "Count", title = "Flow between groupings A and B") +
guides(fill="none") +
scale_fill_manual(values = rep(brewer.pal(12, "Set3"), length.out = length(unique(group_mapping$feature_group))))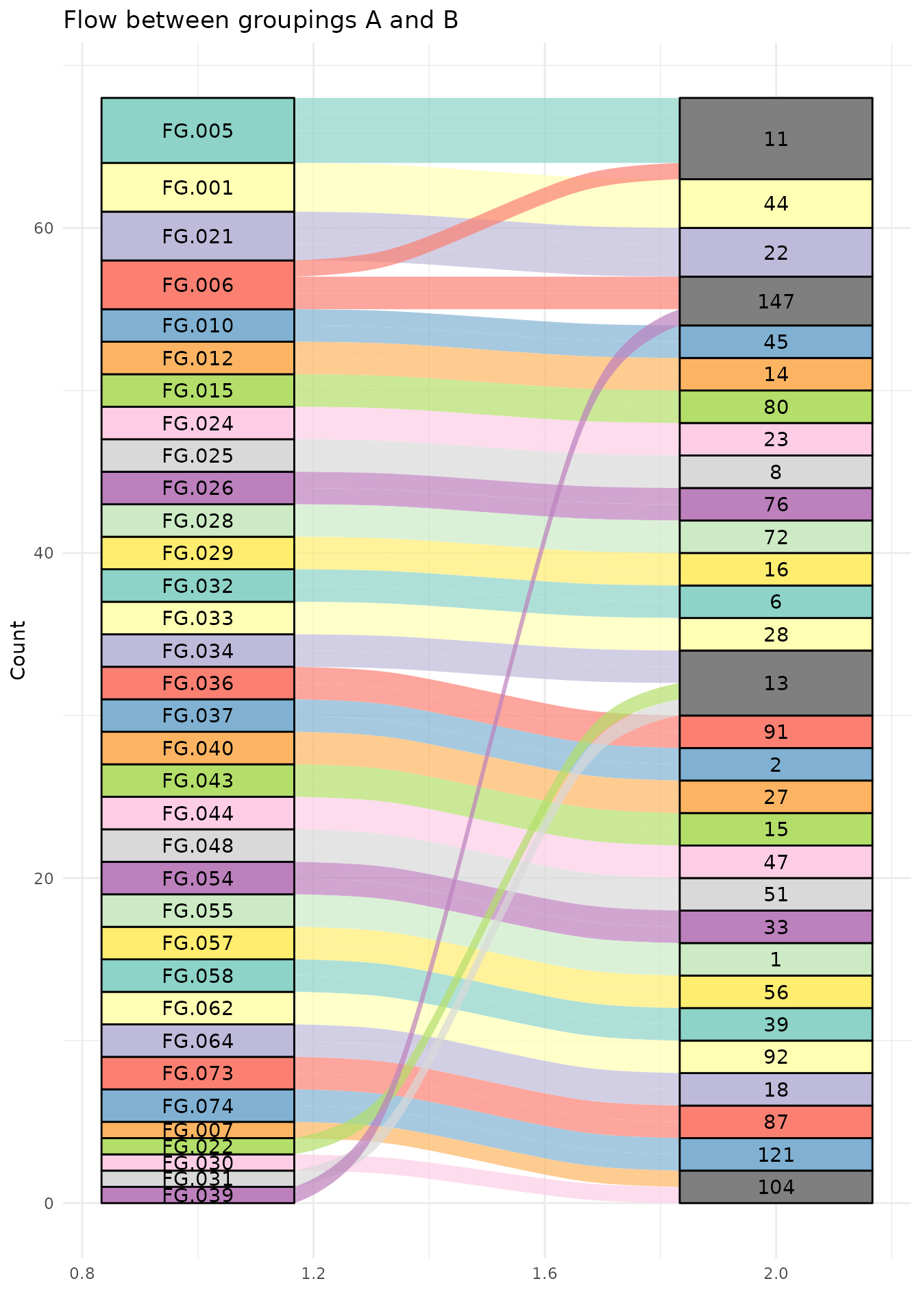
Or just look at those that are not mapped the same by CAMERA and groupFeatures.
group_mapping_different <- group_mapping %>%
group_by(feature_group) %>%
mutate(n_pc = n_distinct(pcgroup)) %>%
group_by(pcgroup) %>%
mutate(n_fg = n_distinct(feature_group)) %>%
ungroup() %>%
filter(n_pc > 1 | n_fg > 1)
ggplot(group_mapping_different, aes(axis1 = feature_group, axis2 = pcgroup, fill = feature_group)) +
geom_alluvium(alpha = 0.7) +
geom_stratum() +
geom_text(stat = "stratum", aes(label = after_stat(stratum))) +
theme_minimal() +
labs(x = "", y = "Count", title = "Flow between groupings A and B") +
guides(fill="none") +
scale_fill_manual(values = rep(brewer.pal(12, "Set3"), length.out = length(unique(group_mapping_different$feature_group))))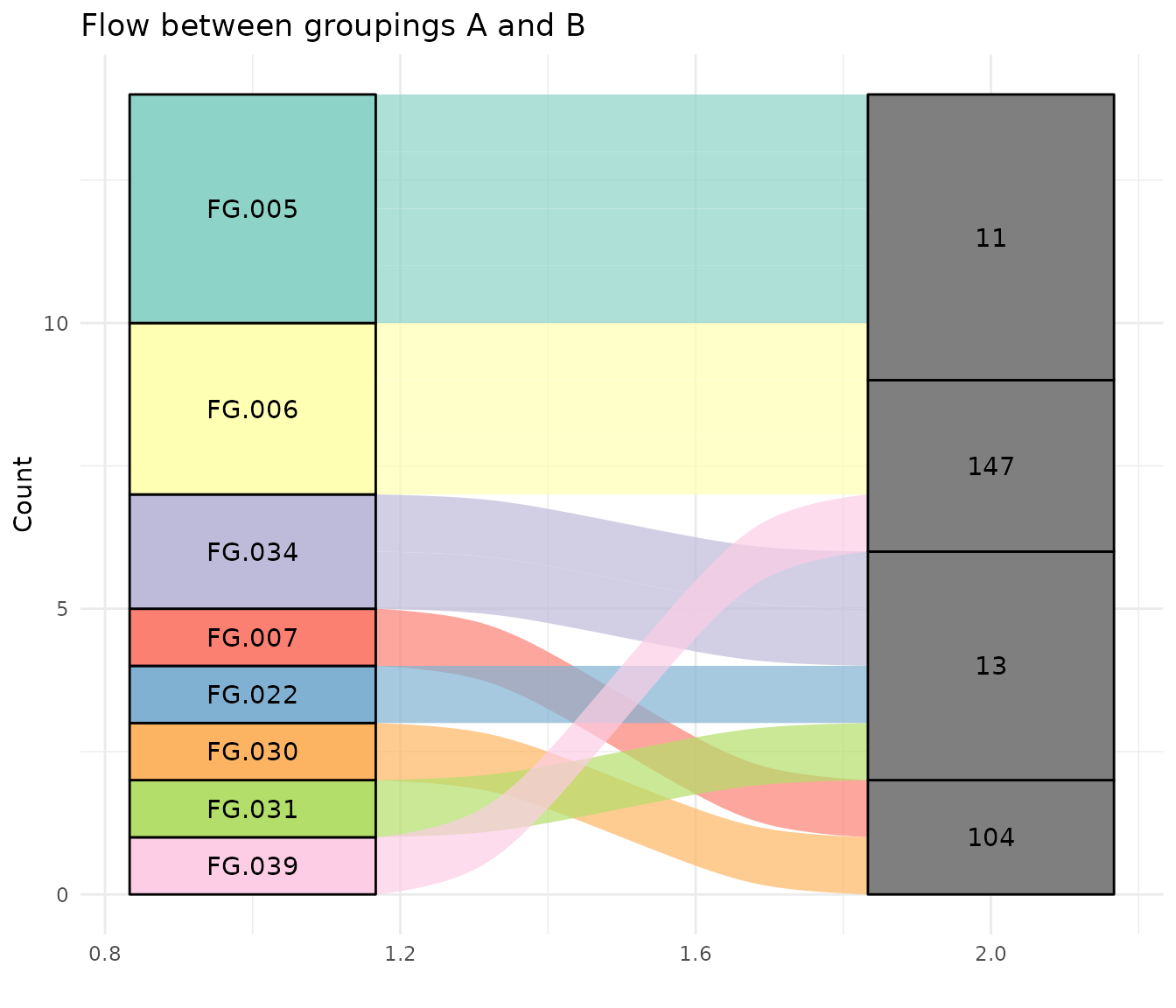
Understanding the Output
The resulting tibble contains comprehensive information about each feature in each sample. Below is a complete description of all columns:
Column Reference Table
The table has been organized into logical groups for easier understanding:
| Column | Content |
|---|---|
| Sample information | |
filepath |
Path to the raw data file |
filename |
The filename without path |
fromFile |
The file number (the order files were supplied in) |
Plus any columns from sampleData() |
Sample metadata columns (e.g., sample_name, sample_group, sample_type) |
| Feature identifiers | |
feature_id |
The index of the feature after grouping across samples |
peakidx |
The index of the peak before grouping across samples |
| Feature-level m/z statistics (across all samples) | |
f_mzmed |
The median m/z found for that feature across samples |
f_mzmin |
The minimum m/z found for that feature across samples |
f_mzmax |
The maximum m/z found for that feature across samples |
| Feature-level retention time statistics (across all samples) | |
f_rtmed |
The median retention time found for that feature across samples |
f_rtmin |
The minimum retention time found for that feature across samples |
f_rtmax |
The maximum retention time found for that feature across samples |
| CAMERA annotations | |
isotopes |
The isotope annotation from CAMERA (e.g., “[M]+”, “[M+1]+”, “[M+2]+”) |
adduct |
The adduct annotation from CAMERA (e.g., “[M+H]+”, “[M+Na]+”, “[M+NH4]+”) |
pcgroup |
The feature grouping index from CAMERA - features with same ID likely come from the same compound |
| MsFeatures annotations | |
feature_group |
The feature group ID from MsFeatures (e.g., “FG.001”, “FG.002”) - features with same ID are grouped by retention time, abundance correlation, or EIC similarity |
| Peak-level m/z measurements (in that specific sample) | |
mz |
The median m/z found for that feature in that sample |
mzmin |
The minimum m/z found for that feature in that sample |
mzmax |
The maximum m/z found for that feature in that sample |
| Peak-level retention time measurements (in that specific sample) | |
rt |
The retention time found for that feature in that sample |
rtmin |
The minimum retention time found for that feature in that sample |
rtmax |
The maximum retention time found for that feature in that sample |
| Peak intensity measurements | |
into |
The area under the peak |
intb |
The area under the peak after baseline removal |
maxo |
The maximum intensity (i.e., height) of the peak |
sn |
The signal to noise ratio of that peak |
| Gaussian peak fitting parameters | |
egauss |
RMSE of Gaussian fit |
mu |
Gaussian parameter mu (center of the Gaussian; unit is scan number) |
sigma |
Gaussian parameter sigma |
h |
Gaussian parameter h (height of the Gaussian peak) |
| CentWave algorithm parameters | |
f |
Region number of m/z ROI where the peak was localized |
dppm |
m/z deviation of mass trace across scans in ppm |
scale |
Scale on which the peak was localized |
scpos |
Center of peak position found by wavelet analysis |
scmin |
Left peak limit found by wavelet analysis (scan number) |
scmax |
Right peak limit found by wavelet analysis (scan number) |
| Additional columns | |
ms_level |
MS level (e.g., MS1, MS2) |
is_filled |
Was the intensity found by gap filling (TRUE) or peak picking (FALSE) |
Important notes:
- Each row represents one feature in one sample
- If a feature was not detected in a sample, peak-level columns (mz,
rt, into, etc.) will be
NA - Feature-level statistics (f_mzmed, f_rtmed, etc.) are always present and represent values across all samples
- CAMERA annotations (isotopes, adduct, pcgroup) are optional and only present when xsAnnotate is provided
- MsFeatures annotations (feature_group) are optional and only present when groupFeatures() was applied
- Both CAMERA and MsFeatures annotations apply at the feature level and are the same across all samples for a given feature
- The distinction between “feature-level” and “peak-level” is
important:
-
Feature-level (prefix
f_): Statistics aggregated across all samples - Peak-level (no prefix): Values for the specific peak in that specific sample
-
Feature-level (prefix
CAMERA Annotations
When CAMERA annotations are included, these additional columns are available:
-
isotopes: Isotope annotation (e.g., “[M]+”, “[M+1]+”) -
adduct: Adduct annotation (e.g., “[M+H]+”, “[M+Na]+”) -
pcgroup: Pseudospectrum correlation group ID
# View features with adduct annotations (using the CAMERA-annotated table)
peak_table_camera %>%
filter(adduct!="") %>%
select(feature_id, f_mzmed, f_rtmed, isotopes, adduct, pcgroup) %>%
distinct(feature_id, .keep_all = TRUE) %>%
head()
#> # A tibble: 6 × 6
#> feature_id f_mzmed f_rtmed isotopes adduct pcgroup
#> <int> <dbl> <dbl> <chr> <chr> <int>
#> 1 3 206 2789. "" [M+2K]2+ 334.047 [M+H-NH3-CO2-C5… 11
#> 2 6 241. 3683. "" [M+H-S]+ 272.112 [M+H-CH3OH]+ 27… 87
#> 3 11 255. 3682. "" [M+H-H2O]+ 272.112 87
#> 4 13 266. 3669. "" [M+H-C2H4-CO2]+ 337.215 [M+H-C4H… 44
#> 5 19 279 2788. "" [M+H-C3H4O]+ 334.047 [M+H-C4H8]+… 11
#> 6 21 281. 3669. "[1][M]+" [M+H-NH3-C3H4]+ 337.215 44Peak-Level Information
For each feature in each sample:
-
mz,rt: Detected peak position -
into: Integrated peak intensity -
intb: Baseline-corrected intensity -
maxo: Maximum intensity -
sn: Signal-to-noise ratio
# View detected peaks (non-NA intensities)
peak_table %>%
filter(!is.na(into)) %>%
select(feature_id, filename, mz, rt, into, sn) %>%
head()
#> # A tibble: 6 × 6
#> feature_id filename mz rt into sn
#> <int> <chr> <dbl> <dbl> <dbl> <dbl>
#> 1 1 ko15.CDF 200. 2933. 135162. NA
#> 2 2 ko15.CDF 205 2792. 1924712. 64
#> 3 3 ko15.CDF 206 2790. 213659. 14
#> 4 4 ko15.CDF 207. 2718. 349011. 17
#> 5 5 ko15.CDF 233 3035. 286221. 23
#> 6 6 ko15.CDF 241. 3681. 1160580. 11Sample Information
-
filename,filepath: Sample file information -
fromFile: Sample index - Plus any columns from
sampleData()(sample metadata)
peak_table %>%
select(feature_id, filename, sample_name, sample_group, sample_type) %>%
head()
#> # A tibble: 6 × 5
#> feature_id filename sample_name sample_group sample_type
#> <int> <chr> <chr> <chr> <chr>
#> 1 1 ko15.CDF ko15 KO QC
#> 2 2 ko15.CDF ko15 KO QC
#> 3 3 ko15.CDF ko15 KO QC
#> 4 4 ko15.CDF ko15 KO QC
#> 5 5 ko15.CDF ko15 KO QC
#> 6 6 ko15.CDF ko15 KO QCMissing Values
Features not detected in a sample have NA for peak-level
columns:
# Count for each feature how many samples the peak was found in
detection_rate <- peak_table %>%
group_by(feature_id, f_mzmed, f_rtmed) %>%
summarise(
n_samples_detected = sum(!is.na(into)),
n_samples_total = n(),
detection_rate = n_samples_detected / n_samples_total,
.groups = "drop"
) %>%
arrange(desc(detection_rate))
hist(detection_rate$detection_rate, breaks=10, main = "Histogram of feature detection rate")
Downstream Analysis Examples
Visualize Feature Intensities
# Plot intensity distribution by sample group
peak_table %>%
filter(feature_id == 10) %>%
ggplot(aes(x = sample_group, y = log10(into), fill = sample_group)) +
geom_boxplot() +
labs(
title = "Feature Intensity Distribution",
x = "Sample Group",
y = "log10(Intensity)"
) +
theme_minimal()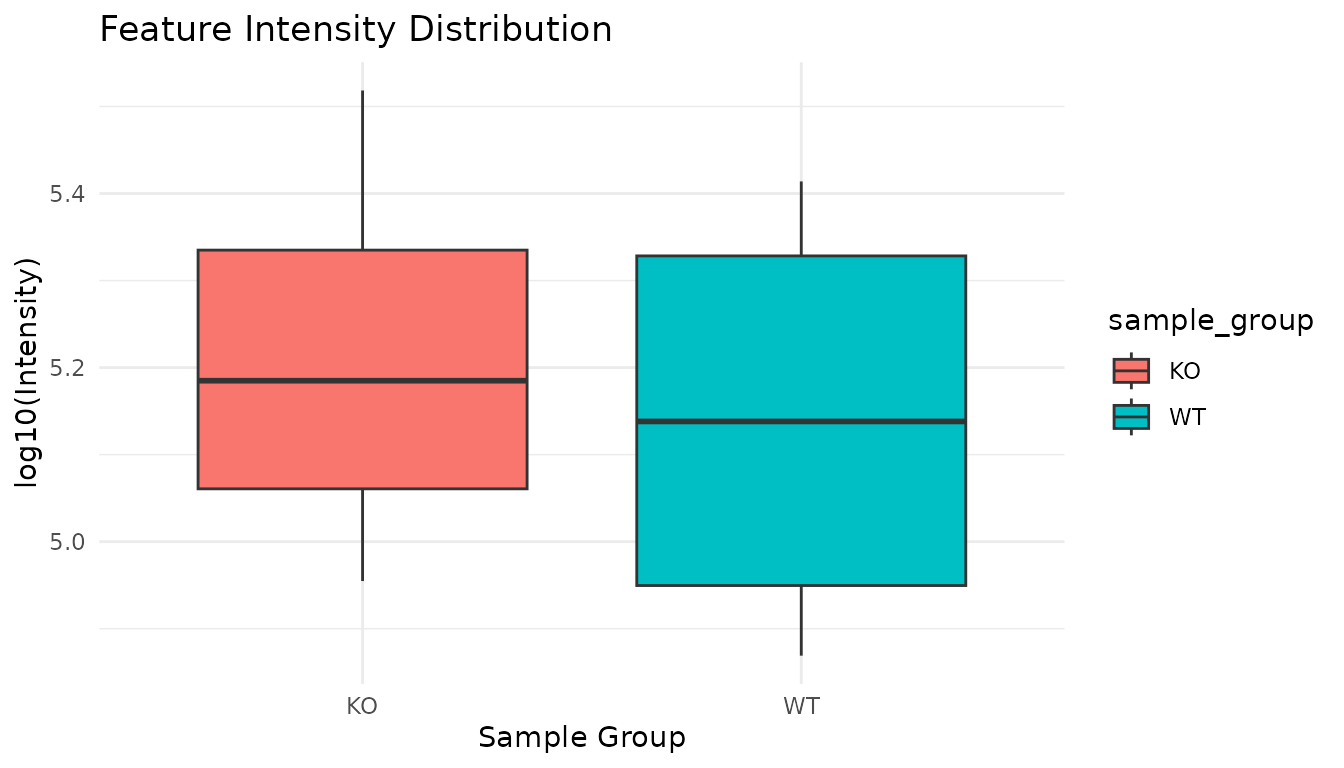
Identify Features with Adduct Annotations
# Count features by adduct type (using the CAMERA-annotated table)
adduct_counts <- peak_table_camera %>%
filter(adduct!="") %>%
separate_rows(adduct, sep = "\\s(?=\\[M)") %>%
distinct(feature_id, adduct) %>%
mutate(adduct = gsub("^(\\[.*\\].*\\+).*","\\1",adduct)) %>%
count(adduct, sort = TRUE)
adduct_counts
#> # A tibble: 33 × 2
#> adduct n
#> <chr> <int>
#> 1 [M+H]+ 12
#> 2 [M+Na]+ 12
#> 3 [M+H-H2O]+ 10
#> 4 [M+K]+ 6
#> 5 [M+H-C3H9N-C2H4O2]+ 5
#> 6 [M+H-CH4]+ 5
#> 7 [M+H-O]+ 5
#> 8 [M+H-NH3]+ 4
#> 9 [M+H-CH3OH]+ 3
#> 10 [M+H-S]+ 3
#> # ℹ 23 more rowsVisualize Adduct Distribution
We can create a pie chart showing the frequency of different adducts and fragments found by CAMERA:
# Create pie chart of adduct frequencies
adduct_counts %>%
mutate(count = n) %>%
ggpie(data = .,
group_key = "adduct",
count_type = "count",
label_info = "group",
label_type = "circle",
label_size = 4,
label_pos = "out"
) +
guides(fill="none") +
ggtitle("Distribution of Adduct Annotations")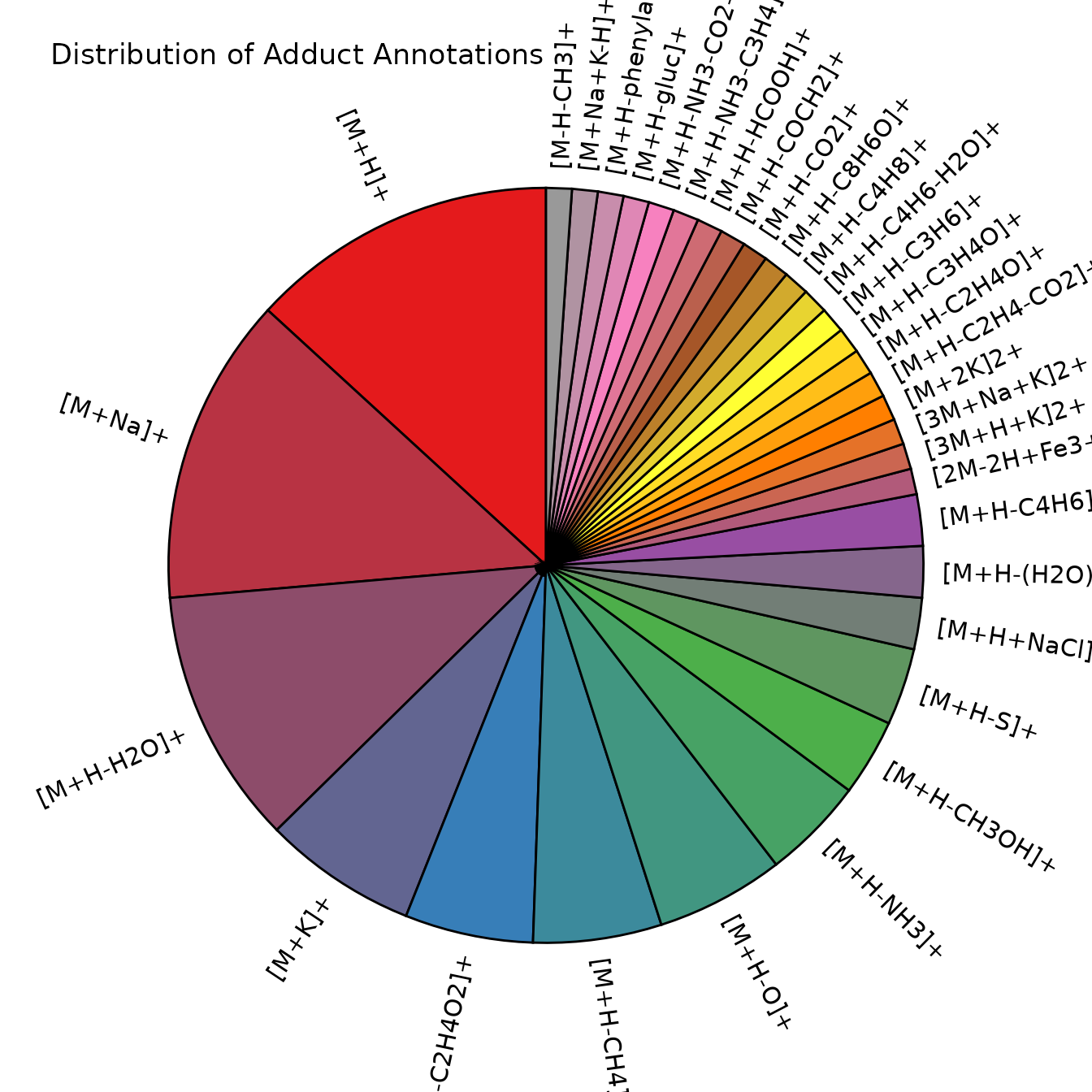
Extract Specific Features for Further Analysis
# Get a specific feature across all samples
feature_123 <- peak_table %>%
filter(feature_id == 123) %>%
select(feature_id, filename, sample_group, mz, rt, into)
head(feature_123)
#> # A tibble: 6 × 6
#> feature_id filename sample_group mz rt into
#> <int> <chr> <chr> <dbl> <dbl> <dbl>
#> 1 123 ko15.CDF KO 371. 3657. 563419.
#> 2 123 ko16.CDF KO 371. 3657. 320434.
#> 3 123 ko21.CDF KO 371. 3649. 123876.
#> 4 123 ko22.CDF KO 371. 3633. 173440.
#> 5 123 wt15.CDF WT 371. 3632. 570454.
#> 6 123 wt16.CDF WT 371. 3639. 287139.Session Info
sessionInfo()
#> R version 4.6.0 (2026-04-24)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] ggpie_0.2.5 RColorBrewer_1.1-3 ggalluvial_0.12.6
#> [4] commonMZ_0.0.2 MsFeatures_1.20.0 CAMERA_1.68.0
#> [7] MSnbase_2.37.0 S4Vectors_0.50.0 mzR_2.46.0
#> [10] Rcpp_1.1.1-1.1 Biobase_2.72.0 BiocGenerics_0.58.0
#> [13] generics_0.1.4 MsExperiment_1.14.0 ProtGenerics_1.44.0
#> [16] tidyr_1.3.2 ggplot2_4.0.3 dplyr_1.2.1
#> [19] xcms_4.10.0 BiocParallel_1.46.0 tidyXCMS_0.99.37
#>
#> loaded via a namespace (and not attached):
#> [1] rstudioapi_0.18.0 jsonlite_2.0.0
#> [3] MultiAssayExperiment_1.38.0 magrittr_2.0.5
#> [5] farver_2.1.2 MALDIquant_1.22.3
#> [7] rmarkdown_2.31 fs_2.1.0
#> [9] ragg_1.5.2 vctrs_0.7.3
#> [11] base64enc_0.1-6 htmltools_0.5.9
#> [13] S4Arrays_1.12.0 progress_1.2.3
#> [15] cellranger_1.1.0 SparseArray_1.12.2
#> [17] Formula_1.2-5 mzID_1.50.0
#> [19] sass_0.4.10 bslib_0.10.0
#> [21] htmlwidgets_1.6.4 desc_1.4.3
#> [23] plyr_1.8.9 impute_1.86.0
#> [25] cachem_1.1.0 igraph_2.3.1
#> [27] lifecycle_1.0.5 iterators_1.0.14
#> [29] pkgconfig_2.0.3 Matrix_1.7-5
#> [31] R6_2.6.1 fastmap_1.2.0
#> [33] MatrixGenerics_1.24.0 clue_0.3-68
#> [35] digest_0.6.39 ggnewscale_0.5.2
#> [37] pcaMethods_2.4.0 colorspace_2.1-2
#> [39] textshaping_1.0.5 Hmisc_5.2-5
#> [41] GenomicRanges_1.64.0 labeling_0.4.3
#> [43] Spectra_1.22.0 abind_1.4-8
#> [45] compiler_4.6.0 bit64_4.8.0
#> [47] withr_3.0.2 doParallel_1.0.17
#> [49] htmlTable_2.5.0 S7_0.2.2
#> [51] backports_1.5.1 PTMods_1.0.0
#> [53] DBI_1.3.0 MASS_7.3-65
#> [55] DelayedArray_0.38.1 tools_4.6.0
#> [57] PSMatch_1.16.0 foreign_0.8-91
#> [59] otel_0.2.0 nnet_7.3-20
#> [61] glue_1.8.1 QFeatures_1.22.0
#> [63] grid_4.6.0 checkmate_2.3.4
#> [65] cluster_2.1.8.2 reshape2_1.4.5
#> [67] gtable_0.3.6 tzdb_0.5.0
#> [69] preprocessCore_1.74.0 data.table_1.18.4
#> [71] hms_1.1.4 MetaboCoreUtils_1.20.1
#> [73] utf8_1.2.6 XVector_0.52.0
#> [75] ggrepel_0.9.8 foreach_1.5.2
#> [77] pillar_1.11.1 stringr_1.6.0
#> [79] vroom_1.7.1 limma_3.68.2
#> [81] lattice_0.22-9 bit_4.6.0
#> [83] tidyselect_1.2.1 RBGL_1.88.0
#> [85] knitr_1.51 gridExtra_2.3
#> [87] IRanges_2.46.0 Seqinfo_1.2.0
#> [89] SummarizedExperiment_1.42.0 xfun_0.57
#> [91] statmod_1.5.1 matrixStats_1.5.0
#> [93] stringi_1.8.7 lazyeval_0.2.3
#> [95] yaml_2.3.12 evaluate_1.0.5
#> [97] codetools_0.2-20 MsCoreUtils_1.24.0
#> [99] tibble_3.3.1 BiocManager_1.30.27
#> [101] graph_1.90.0 cli_3.6.6
#> [103] affyio_1.82.0 rpart_4.1.27
#> [105] systemfonts_1.3.2 jquerylib_0.1.4
#> [107] readxl_1.4.5 MassSpecWavelet_1.78.0
#> [109] XML_3.99-0.23 parallel_4.6.0
#> [111] pkgdown_2.2.0 readr_2.2.0
#> [113] prettyunits_1.2.0 AnnotationFilter_1.36.0
#> [115] scales_1.4.0 affy_1.90.0
#> [117] ncdf4_1.24 purrr_1.2.2
#> [119] crayon_1.5.3 rlang_1.2.0
#> [121] vsn_3.80.0