ggplot2 Version of plot() for XChromatogram
Source: R/AllGenerics.R, R/gplot-LamaParama-methods.R, R/gplot-methods.R
gplot.RdCreates a ggplot2 version of a chromatogram with detected peaks marked.
This is equivalent to the base R plot() method for XChromatogram objects.
Creates a ggplot2 version of the retention time alignment model
visualization for xcms::LamaParama() objects.
LamaParama objects contain parameters and results from landmark-based
retention time alignment.
Usage
gplot(x, ...)
# S4 method for class 'LamaParama'
gplot(x, index = 1L, colPoints = "#00000060", colFit = "#00000080", ...)
# S4 method for class 'XChromatogram'
gplot(
x,
col = "black",
lty = 1,
type = "l",
peakType = c("polygon", "point", "rectangle", "none"),
peakCol = "#00000060",
peakBg = "#00000020",
peakPch = 1,
...
)
# S4 method for class 'XChromatograms'
gplot(
x,
col = "#00000060",
lty = 1,
type = "l",
peakType = c("polygon", "point", "rectangle", "none"),
peakCol = "#00000060",
peakBg = "#00000020",
peakPch = 1,
include_columns = NULL,
...
)
# S4 method for class 'MChromatograms'
gplot(
x,
col = "#00000060",
lty = 1,
type = "l",
peakType = c("polygon", "point", "rectangle", "none"),
peakCol = "#00000060",
peakBg = "#00000020",
peakPch = 1,
include_columns = NULL,
...
)Arguments
- x
A
LamaParamaobject containing retention time alignment parameters and results.- ...
Additional parameters (currently unused, for S4 compatibility).
- index
integer(1)specifying which retention time map to plot (default:1).- colPoints
Color for the matched peak points (default: semi-transparent black).
- colFit
Color for the fitted model line (default: semi-transparent black).
- col
Color for the chromatogram line (default:
"black"). ForXChromatogramsandMChromatogramsobjects, this can also be a column name frompData(x)to map color to sample metadata (e.g.,col = "sample_group"). When a column name is used, ggplot2 automatically adds a color legend.- lty
Line type for chromatogram (default:
1).- type
Plot type (default:
"l"for line).- peakType
character(1)defining the type of peak annotation:"polygon","point","rectangle", or"none"(default:"polygon").- peakCol
Color for peak markers (default:
"#00000060"). ForXChromatogramsobjects, this can also be a column name frompData(x)to map peak border color to sample metadata.- peakBg
Background color for peak markers (default:
"#00000020"). ForXChromatogramsobjects, this can also be a column name frompData(x)to map peak fill color to sample metadata.- peakPch
Point character for peak markers when
peakType = "point"(default:1).- include_columns
For
XChromatogramsandMChromatograms: whichpData(x)columns to include in plotly tooltips.NULL(default) for no metadata in tooltips,TRUEfor all columns, or acharactervector of specific column names. The tooltip text is stored in thetextaesthetic and is used byplotly::ggplotly().
Details
This function creates a complete chromatogram plot with detected peaks
automatically marked, similar to the base R plot() method for
XChromatogram objects. If the chromatogram contains detected peaks,
they will be shown according to the peakType parameter.
Color Mapping for Multi-Sample Chromatograms
When plotting XChromatograms or MChromatograms objects (multiple
samples), the col, peakCol, and peakBg parameters accept either:
A static color string (e.g.,
col = "red",col = "#00000060") — all samples use the same color (default behavior).A column name from
pData(x)as a quoted string (e.g.,col = "sample_group") — each sample is colored according to its metadata value. The column must exist inBiobase::pData(x), which inherits fromsampleData()of the parentXcmsExperimentobject. The value is automatically detected as a column name when it matches a column inpData(x).
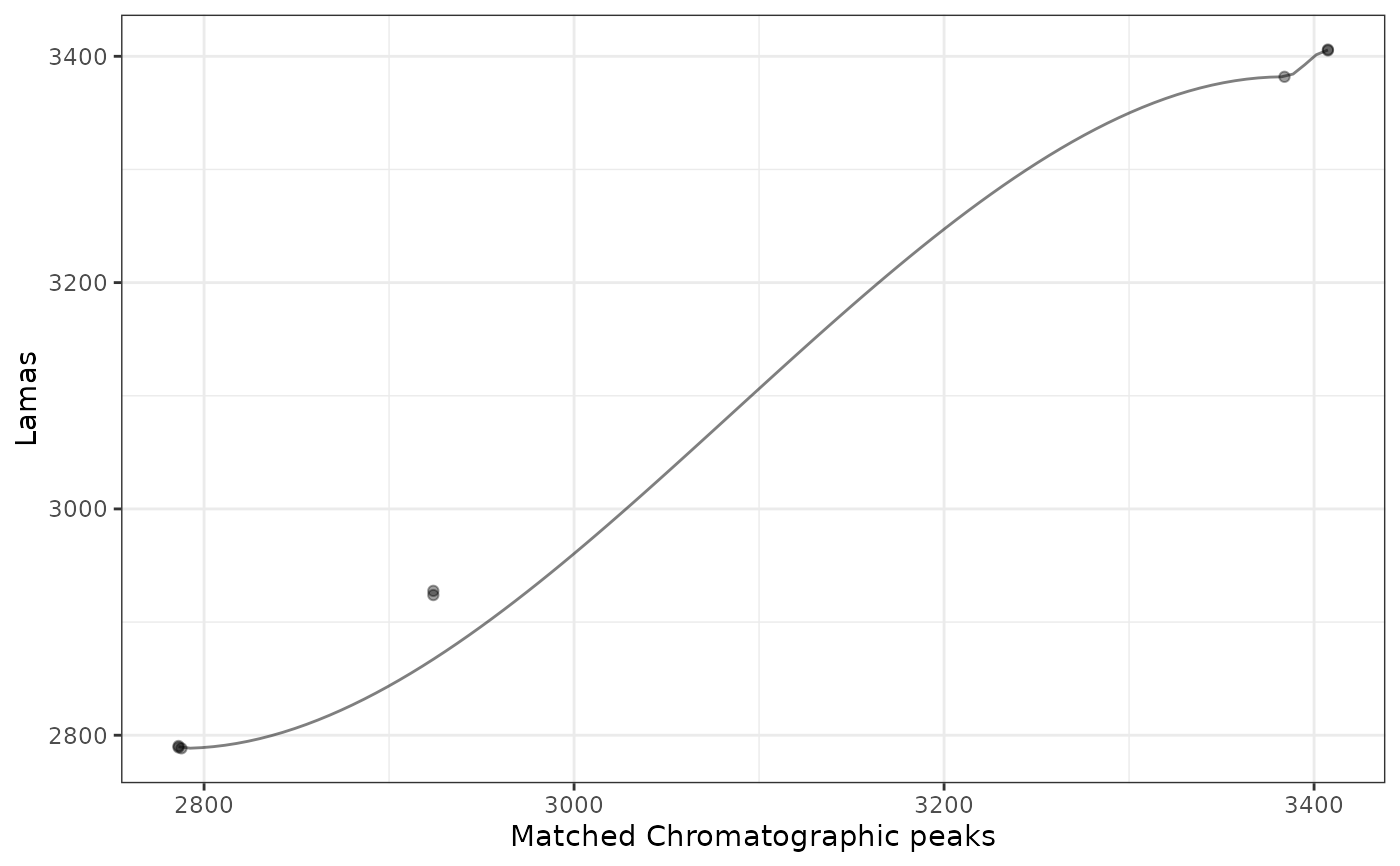
This function visualizes the retention time alignment model for a specific sample.
The plot shows:
Points representing matched chromatographic peaks between the sample and reference
A fitted line (loess or GAM) showing the retention time correction model
The LamaParama object contains parameters for landmark-based alignment
including:
method: The fitting method ("loess" or "gam")span: Span parameter for loess fittingoutlierTolerance: Tolerance for outlier detectionzeroWeight: Weight for the (0,0) anchor pointbs: Basis function for GAM fittingrtMap: List of data frames with retention time pairs
See also
plot,XChromatogram,ANY-method for the original xcms
implementation
Examples
library(xcmsVis)
library(xcms)
library(faahKO)
library(MsExperiment)
library(ggplot2)
## Load preprocessed data
xdata <- loadXcmsData()
# Extract chromatogram
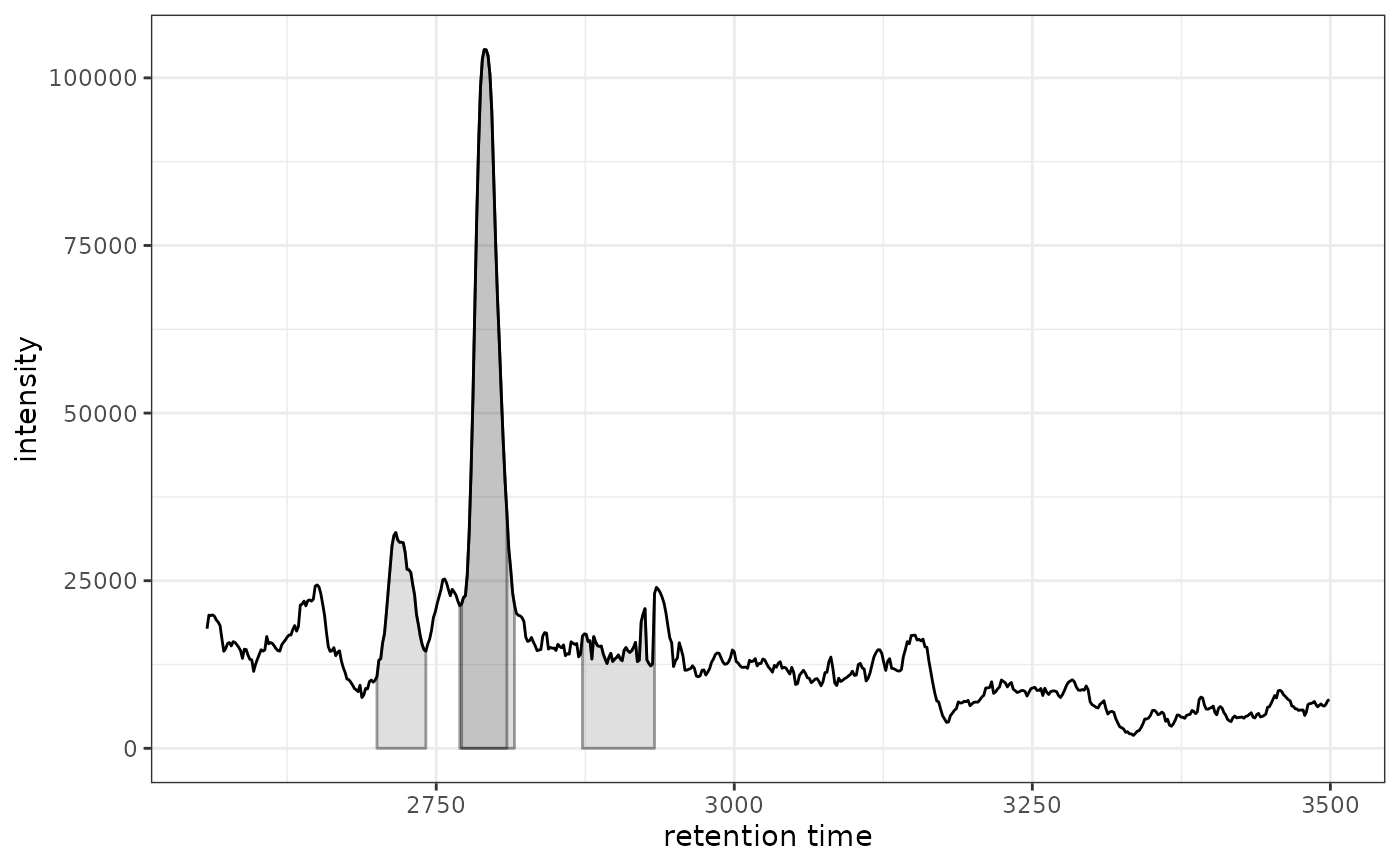
chr <- chromatogram(xdata, mz = c(200, 210), rt = c(2500, 3500))
#> Extracting chromatographic data
#> Processing chromatographic peaks
#> Processing features
# Plot with ggplot2
gplot(chr[1, 1])
#> Warning: Ignoring unknown aesthetics: text
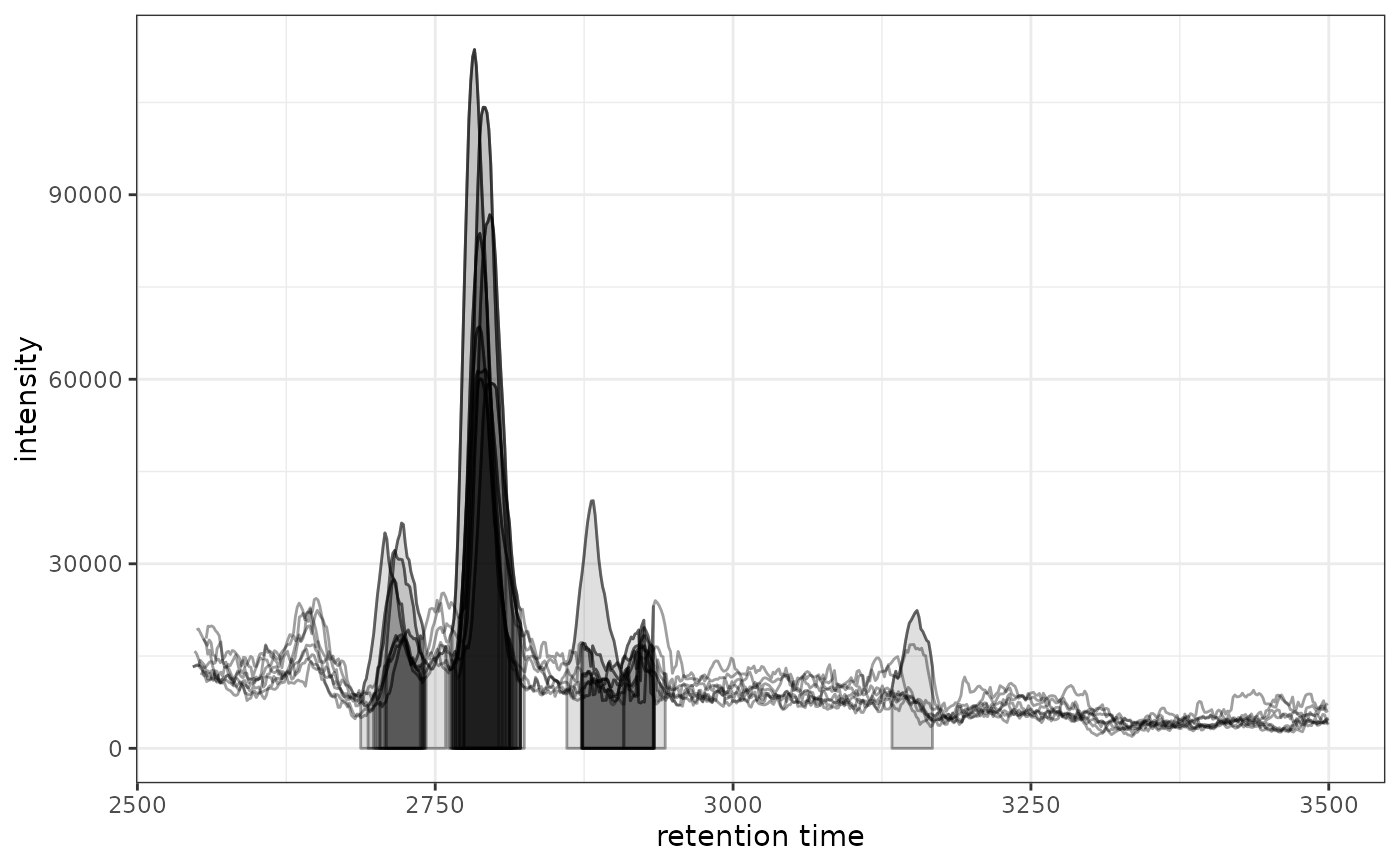
 # Plot data for all samples
gplot(chr)
#> Warning: Ignoring unknown aesthetics: text
# Plot data for all samples
gplot(chr)
#> Warning: Ignoring unknown aesthetics: text
 # Color by sample metadata (requires pData column)
# gplot(chr, col = "sample_group")
library(xcmsVis)
library(xcms)
library(MsExperiment)
# LamaParama requires a reference dataset with landmarks
# See vignette("04-retention-time-alignment") for complete workflow
# Load reference and test datasets
ref <- loadXcmsData("xmse")
tst <- loadXcmsData("faahko_sub2")
# Extract landmarks from QC samples in reference
f <- sampleData(ref)$sample_type
f[f != "QC"] <- NA
ref_filtered <- filterFeatures(
ref, PercentMissingFilter(threshold = 0, f = f))
#> 4 features were removed
ref_mz_rt <- featureDefinitions(ref_filtered)[, c("mzmed", "rtmed")]
# Create and apply LamaParama alignment
lama_param <- LamaParama(lamas = ref_mz_rt, method = "loess", span = 0.5)
tst_adjusted <- adjustRtime(tst, param = lama_param)
# Extract LamaParama result for visualization
proc_hist <- processHistory(tst_adjusted,
type = xcms:::.PROCSTEP.RTIME.CORRECTION)
lama_result <- proc_hist[[length(proc_hist)]]@param
# Visualize the first sample's alignment
gplot(lama_result, index = 1)
#> Warning: pseudoinverse used at -17.037
#> Warning: neighborhood radius 2804.8
#> Warning: reciprocal condition number 0
#> Warning: There are other near singularities as well. 2.5061e+05
#> Warning: span too small. fewer data values than degrees of freedom.
#> Warning: pseudoinverse used at -17.037
#> Warning: neighborhood radius 2803.2
#> Warning: reciprocal condition number 0
#> Warning: There are other near singularities as well. 1641.2
# Color by sample metadata (requires pData column)
# gplot(chr, col = "sample_group")
library(xcmsVis)
library(xcms)
library(MsExperiment)
# LamaParama requires a reference dataset with landmarks
# See vignette("04-retention-time-alignment") for complete workflow
# Load reference and test datasets
ref <- loadXcmsData("xmse")
tst <- loadXcmsData("faahko_sub2")
# Extract landmarks from QC samples in reference
f <- sampleData(ref)$sample_type
f[f != "QC"] <- NA
ref_filtered <- filterFeatures(
ref, PercentMissingFilter(threshold = 0, f = f))
#> 4 features were removed
ref_mz_rt <- featureDefinitions(ref_filtered)[, c("mzmed", "rtmed")]
# Create and apply LamaParama alignment
lama_param <- LamaParama(lamas = ref_mz_rt, method = "loess", span = 0.5)
tst_adjusted <- adjustRtime(tst, param = lama_param)
# Extract LamaParama result for visualization
proc_hist <- processHistory(tst_adjusted,
type = xcms:::.PROCSTEP.RTIME.CORRECTION)
lama_result <- proc_hist[[length(proc_hist)]]@param
# Visualize the first sample's alignment
gplot(lama_result, index = 1)
#> Warning: pseudoinverse used at -17.037
#> Warning: neighborhood radius 2804.8
#> Warning: reciprocal condition number 0
#> Warning: There are other near singularities as well. 2.5061e+05
#> Warning: span too small. fewer data values than degrees of freedom.
#> Warning: pseudoinverse used at -17.037
#> Warning: neighborhood radius 2803.2
#> Warning: reciprocal condition number 0
#> Warning: There are other near singularities as well. 1641.2