library(xcms)
library(xcmsVis)
library(MsExperiment)
library(MsFeatures)
library(ggplot2)
library(BiocParallel)
library(patchwork)
# Configure for serial processing
register(SerialParam())Introduction
This vignette covers the final step in the xcms metabolomics workflow: feature grouping. After retention time alignment, these functions help you:
- Visualize relationships between features
- Identify isotopes, adducts, and fragments
- Assess feature annotation quality
- Create publication-ready feature network plots
xcms Workflow Context
┌───────────────────────────────┐
│ 1. Raw Data Visualization │
│ 2. Peak Detection │
│ 3. Peak Correspondence │
│ 4. Retention Time Alignment │
├───────────────────────────────┤
│ 5. FEATURE GROUPING │ ← YOU ARE HERE
└───────────────────────────────┘What is Feature Grouping?
Feature grouping identifies features that likely represent the same compound. After feature detection and correspondence, groupFeatures() connects features that may be:
- Isotopes: M+1, M+2 isotopic peaks
- Adducts: [M+H]+, [M+Na]+, [M+K]+, etc.
- Fragments: In-source fragmentation products
- Correlated features: Compounds with similar abundance patterns
The gplotFeatureGroups() function visualizes these relationships by plotting features connected by lines within each group across retention time and m/z dimensions.
Functions Covered
| Function | Purpose | Input |
|---|---|---|
gplotFeatureGroups() |
Visualize feature group relationships |
XcmsExperiment, XCMSnExp
|
Setup
Load and Process Data
Feature grouping requires a complete xcms workflow: peak detection, correspondence, retention time alignment, re-correspondence, and then feature grouping.
We’ll use pre-processed data with peaks already detected:
# Load pre-processed data with detected peaks
# This dataset contains 248 detected peaks from 3 samples
xdata <- loadXcmsData("faahko_sub2")
# Add sample metadata
sampleData(xdata)$sample_name <- c("KO01", "KO02", "WT01")
sampleData(xdata)$sample_group <- c("KO", "KO", "WT")
cat("Loaded", length(fileNames(xdata)), "files\n")
#> Loaded 3 files
cat("Detected peaks:", nrow(chromPeaks(xdata)), "\n")
#> Detected peaks: 248Complete XCMS Workflow
# 1. Peak grouping (correspondence)
pdp <- PeakDensityParam(sampleGroups = sampleData(xdata)$sample_group,
minFraction = 0.5, bw = 30)
xdata <- groupChromPeaks(xdata, param = pdp)
cat("Grouped into", nrow(featureDefinitions(xdata)), "features\n")
#> Grouped into 152 features
# 2. Retention time alignment
xdata <- adjustRtime(xdata, param = ObiwarpParam())
# 3. Re-group after alignment
xdata <- groupChromPeaks(xdata, param = pdp)
cat("After alignment:", nrow(featureDefinitions(xdata)), "features\n")
#> After alignment: 152 features
# 4. Group features (identify related features)
xdata <- groupFeatures(xdata, param = SimilarRtimeParam(diffRt = 20))
cat("Identified", length(unique(featureGroups(xdata))), "feature groups\n")
#> Identified 63 feature groupsBasic Feature Group Visualization
The default plot shows all feature groups, with features connected by lines within each group:
gplotFeatureGroups(xdata)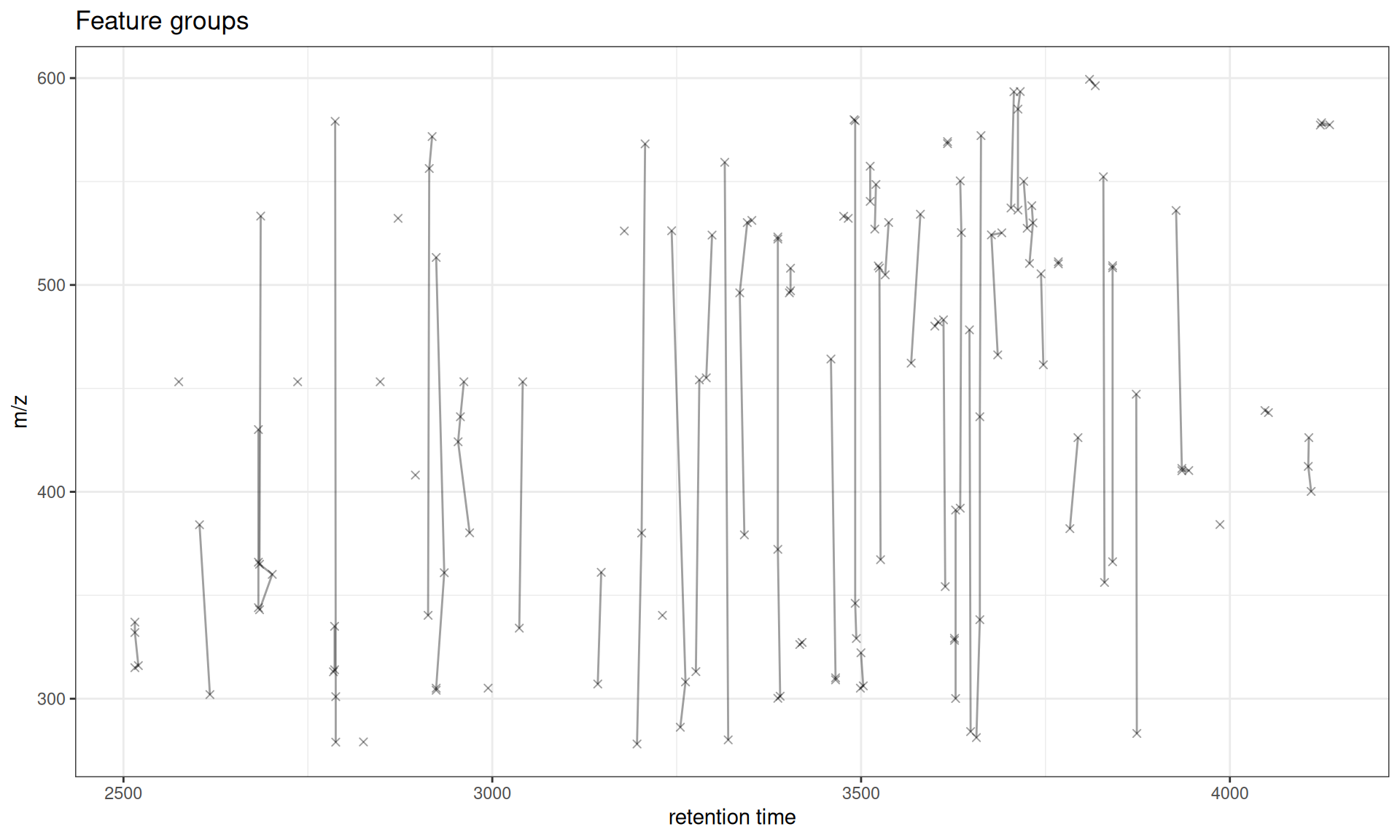
Interpretation
- Points: Individual features (at their median RT and m/z across samples)
- Lines: Connect features within the same group
- Groups: Represent features likely from the same compound (isotopes, adducts, etc.)
Filtering to Specific Feature Groups
You can visualize specific feature groups of interest:
# Get all feature group IDs
all_groups <- unique(featureGroups(xdata))
cat("Feature groups:", head(all_groups, 10), "\n")
#> Feature groups: FG.043 FG.012 FG.054 FG.038 FG.019 FG.023 FG.028 FG.048 FG.009 FG.018
# Plot first 5 groups
gplotFeatureGroups(xdata, featureGroups = all_groups[1:5]) +
ggtitle("First 5 Feature Groups")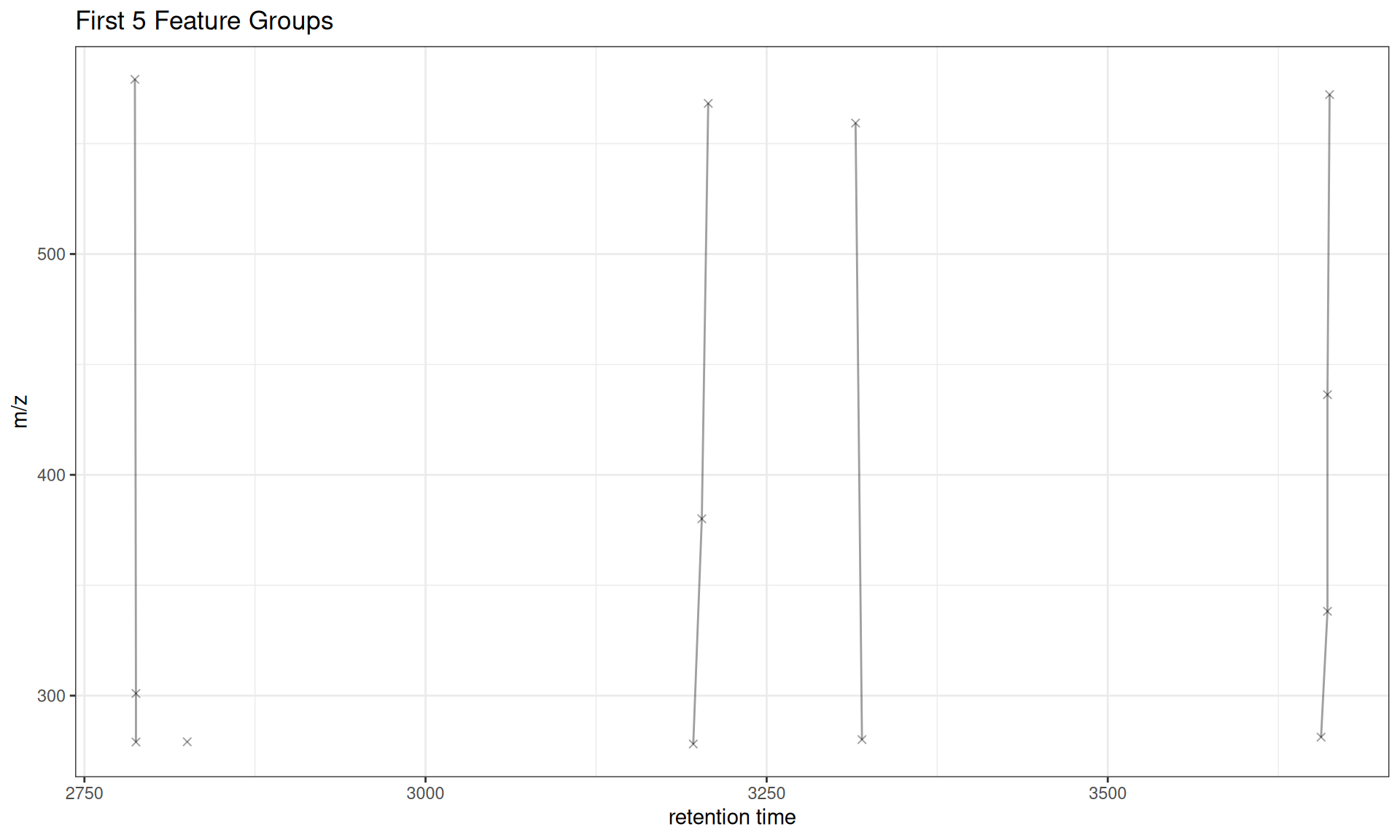
Customization
Custom Styling
# Get first 5 feature groups for clearer visualization
all_groups <- unique(featureGroups(xdata))
gplotFeatureGroups(xdata,
featureGroups = all_groups[1:5],
col = "#E31A1C", # Red color
pch = 16) + # Solid circles
labs(x = "Retention Time (sec)",
y = "Mass-to-Charge Ratio (m/z)",
title = "Custom Styled Feature Groups")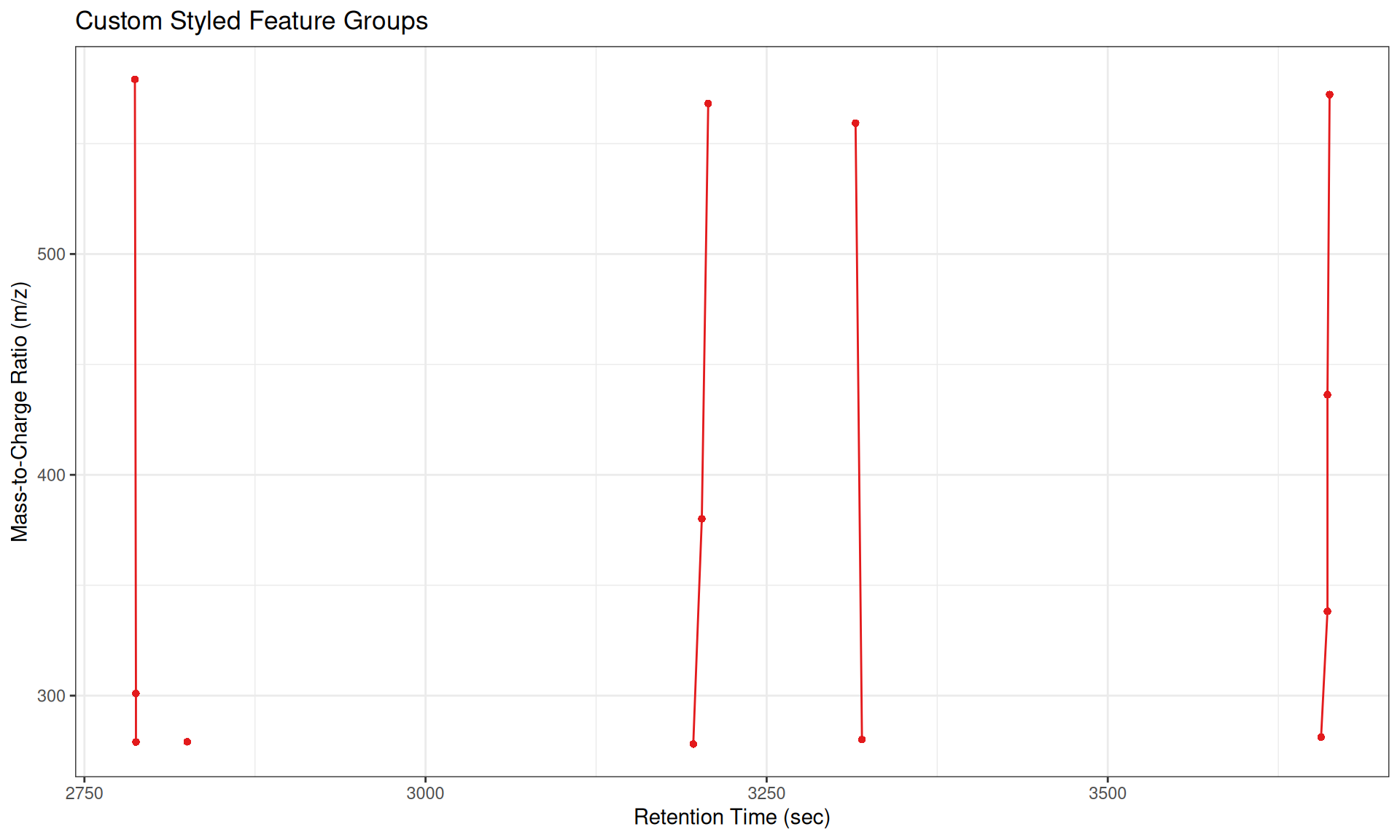
Different Plot Types
The type parameter controls whether to show lines, points, or both:
# Use subset of feature groups for clearer visualization
fg_subset <- all_groups[1:5]
# Plot with lines and points (default)
p1 <- gplotFeatureGroups(xdata, featureGroups = fg_subset, type = "o") +
ggtitle('type = "o" (overplot - lines + points)')
# Plot with lines only
p2 <- gplotFeatureGroups(xdata, featureGroups = fg_subset, type = "l") +
ggtitle('type = "l" (lines only)')
# Plot with points only
p3 <- gplotFeatureGroups(xdata, featureGroups = fg_subset, type = "p", pch = 16) +
ggtitle('type = "p" (points only)')
# Combine plots
p1 / p2 / p3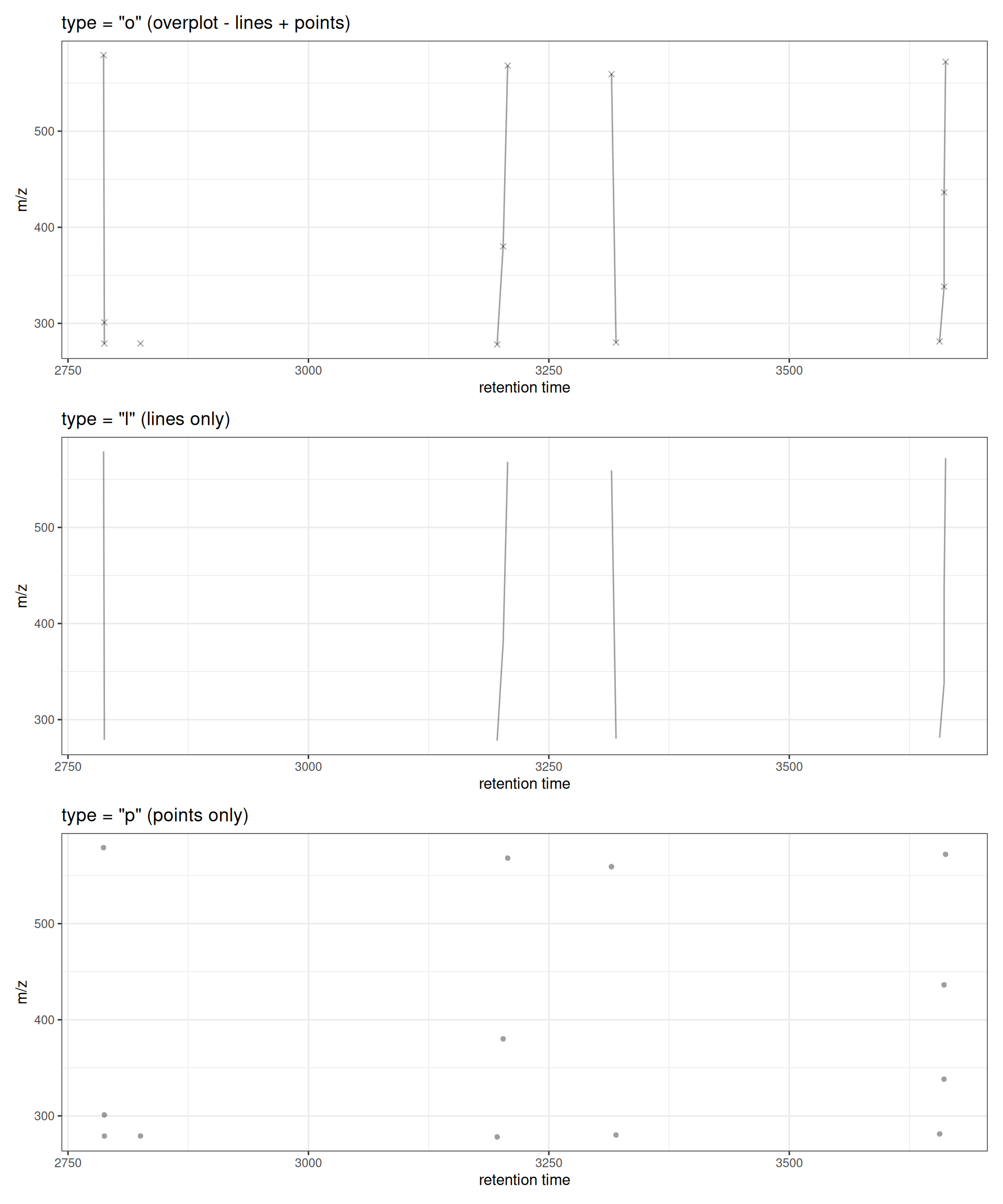
Zooming to Specific Regions
Use xlim and ylim to focus on specific retention time or m/z ranges:
# Focus on features between 3200-3300 seconds RT and specific feature groups
gplotFeatureGroups(xdata,
featureGroups = fg_subset,
xlim = c(3100, 3400),
ylim = c(200, 600)) +
ggtitle("Features in RT 3200-3300 sec, m/z 200-600")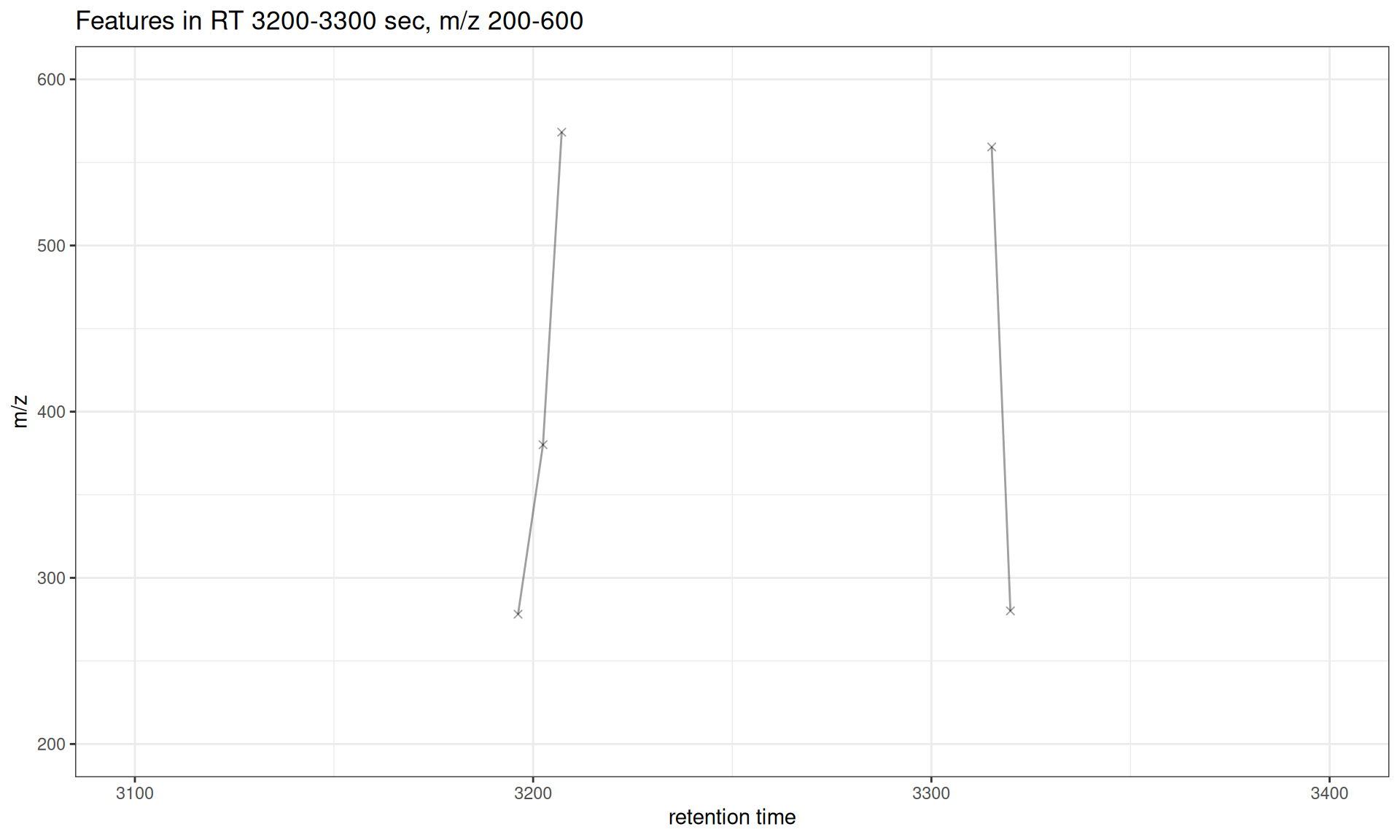
Interactive Visualization
Convert to interactive plotly plot for exploration:
library(plotly)
# Use subset for better interactivity
p <- gplotFeatureGroups(xdata, featureGroups = fg_subset)
ggplotly(p)Understanding Feature Groups
Feature groups represent features that are likely derived from the same compound. Common grouping parameters:
Similar Retention Time
# Group features with similar retention times (likely isotopes/adducts)
xdata_rt <- groupFeatures(xdata, param = SimilarRtimeParam(diffRt = 10))
cat("SimilarRtimeParam (diffRt=10):",
length(unique(featureGroups(xdata_rt))), "groups\n")
#> SimilarRtimeParam (diffRt=10): 76 groups
# Show first 5 groups
fg_rt <- unique(featureGroups(xdata_rt))
gplotFeatureGroups(xdata_rt, featureGroups = fg_rt[1:5]) +
ggtitle("Feature Groups: Similar Retention Time (diffRt = 10 sec)")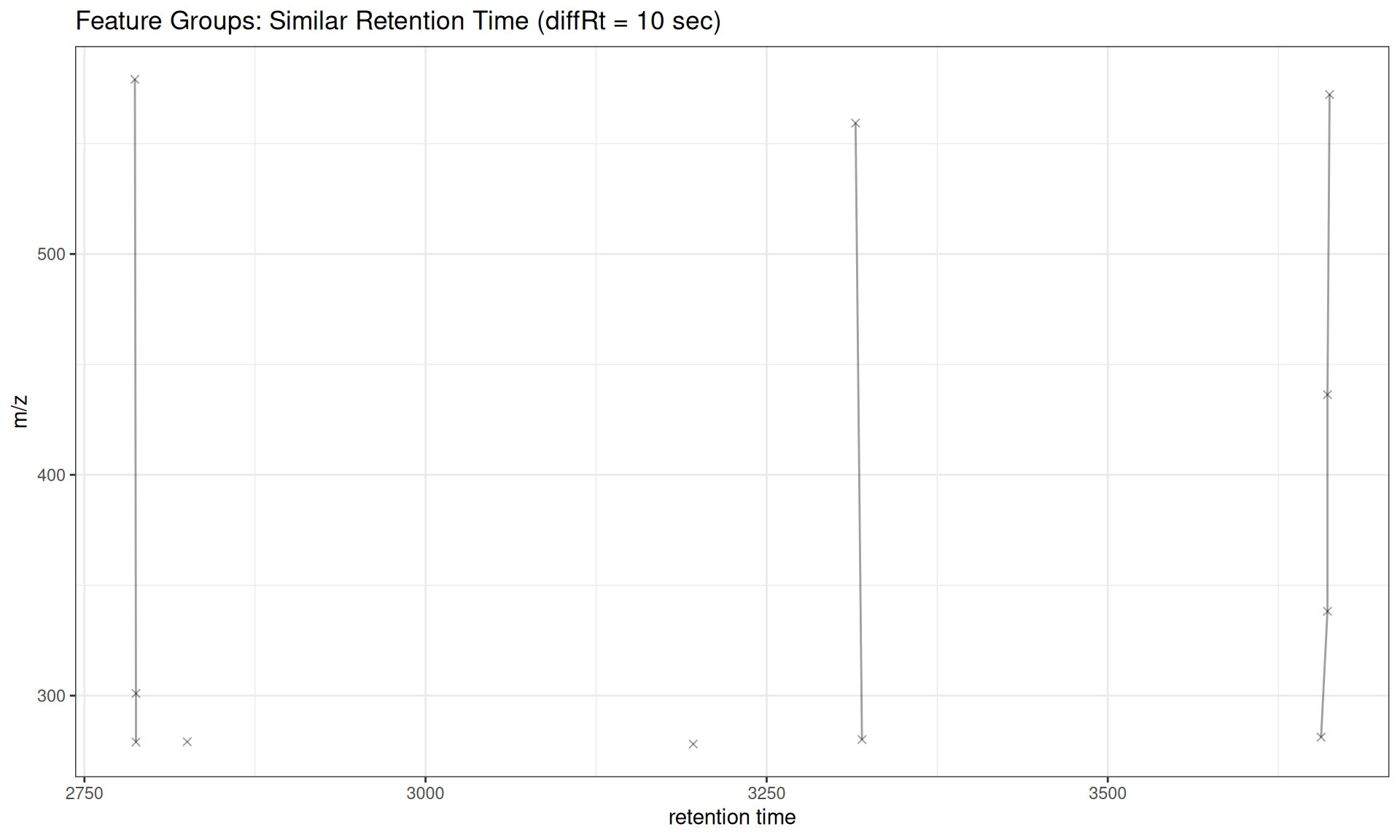
Abundance Correlation
# Group features with correlated abundances across samples
xdata_cor <- groupFeatures(
xdata, param = AbundanceSimilarityParam(threshold = 0.7))
cat("AbundanceSimilarityParam (threshold=0.7):",
length(unique(featureGroups(xdata_cor))), "groups\n")
#> AbundanceSimilarityParam (threshold=0.7): 139 groups
# Show some groups with multiple Features
fg_cor <- names(rev(sort(table(featureGroups(xdata_cor)))))
gplotFeatureGroups(xdata_cor, featureGroups = fg_cor[1:5]) +
ggtitle("Feature Groups: Abundance Correlation (threshold = 0.7)")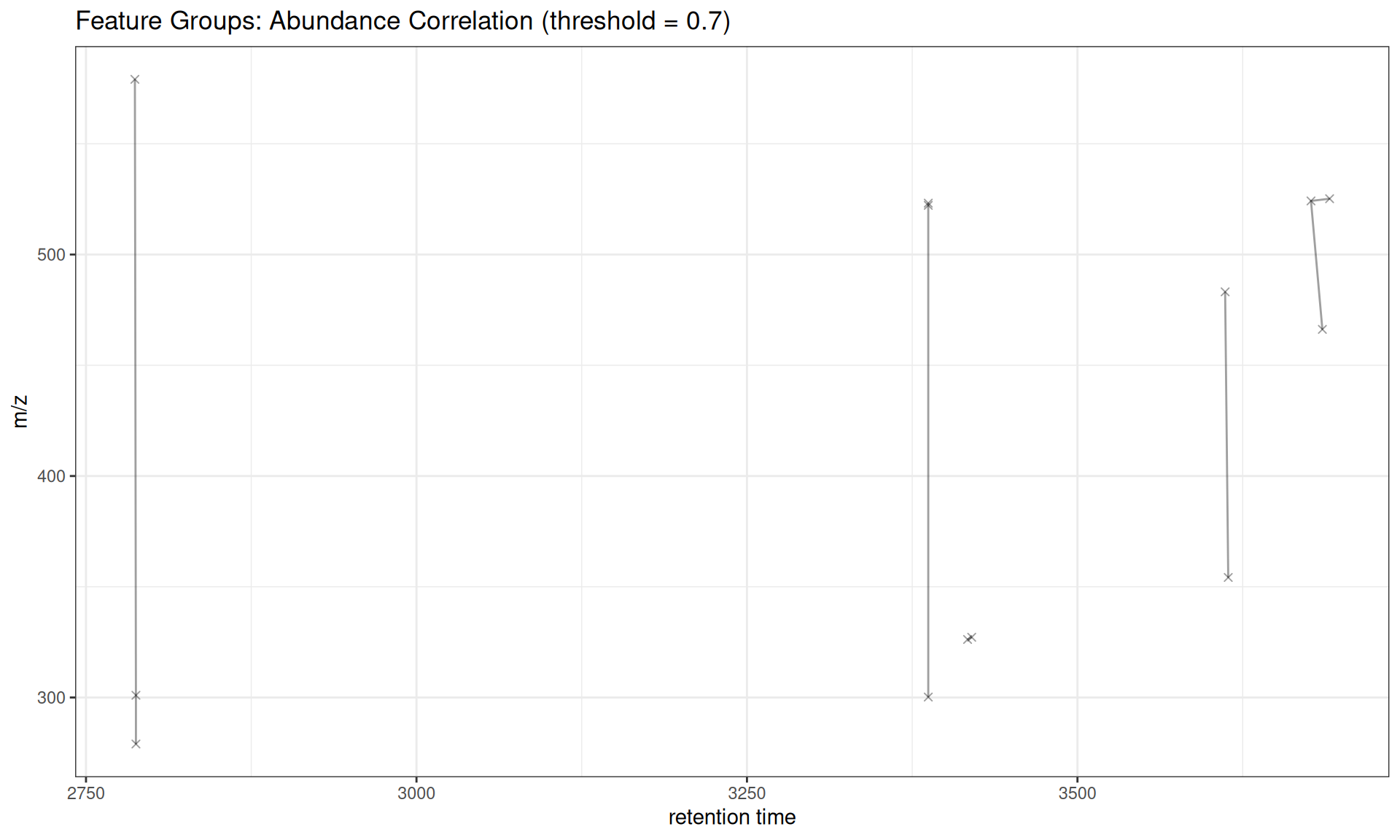
Use Cases
Isotope Pattern Identification
Features grouped by similar RT and abundance correlation may represent isotope patterns (e.g. M, M+1, M+2) or adducts (e.g. [M+H]+, [M+Na]+).
Adduct Identification
Features with similar RT but different m/z values that correlate in abundance may be different adducts of the same compound.
Quality Control
Visualize feature groups to:
- Verify grouping parameters are appropriate
- Identify over-grouping (too many features in one group)
- Identify under-grouping (features that should be grouped but aren’t)
Summary
Use Cases
- Compound annotation: Identify isotopes, adducts, and fragments
- Quality control: Verify feature grouping quality
- Method development: Optimize groupFeatures parameters
- Publication: Create network plots of feature relationships
Workflow Complete!
You’ve now completed the full xcms visualization workflow:
✓ 1. Raw Data Visualization
✓ 2. Peak Detection
✓ 3. Peak Correspondence
✓ 4. Retention Time Alignment
✓ 5. Feature GroupingComparison with Original xmcs
Original xcms Version
# Get first 5 feature groups for comparison
fg_compare <- unique(featureGroups(xdata))[1:5]
# XCMS original (base R graphics)
plotFeatureGroups(xdata, featureGroups = fg_compare)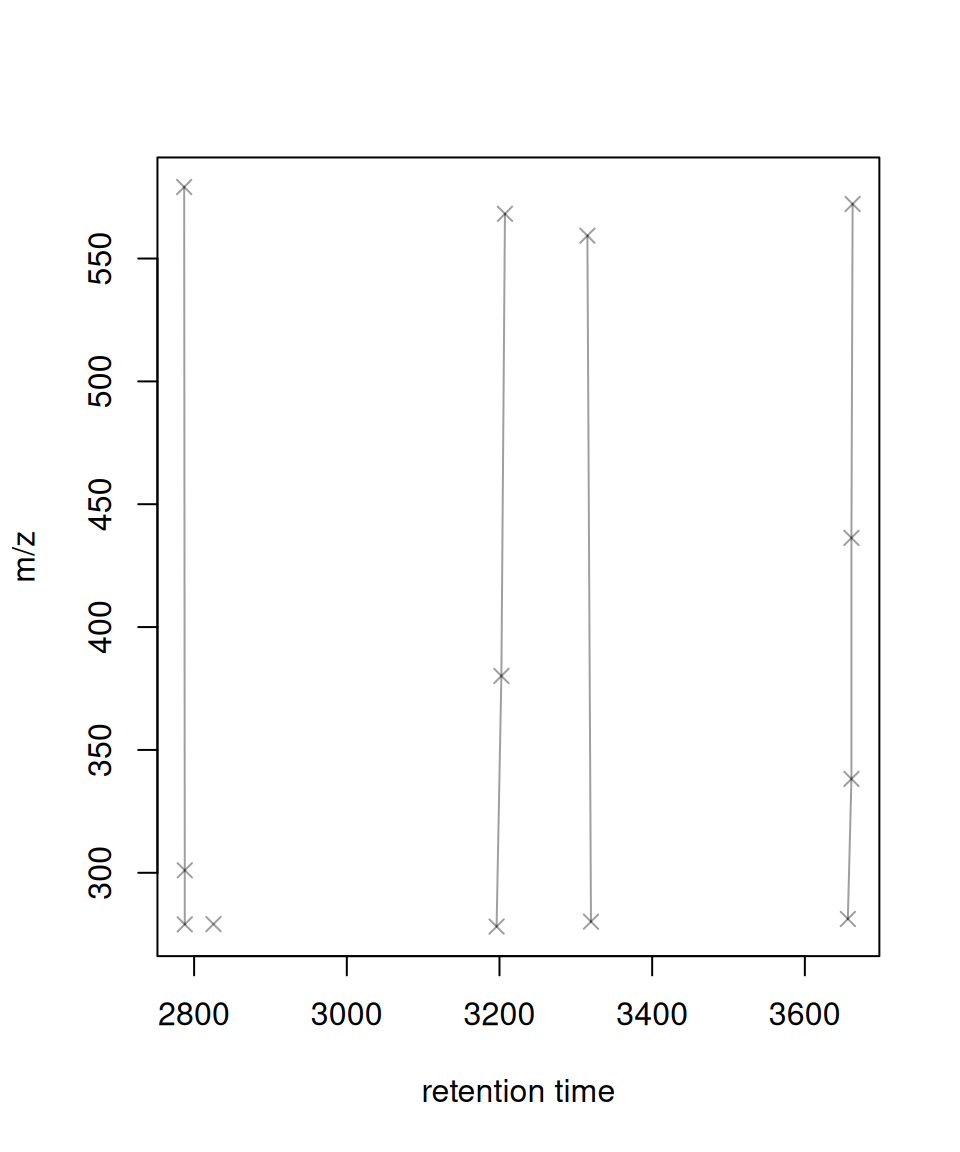
xcmsVis ggplot2 Version
# xcmsVis version (ggplot2)
gplotFeatureGroups(xdata, featureGroups = fg_compare)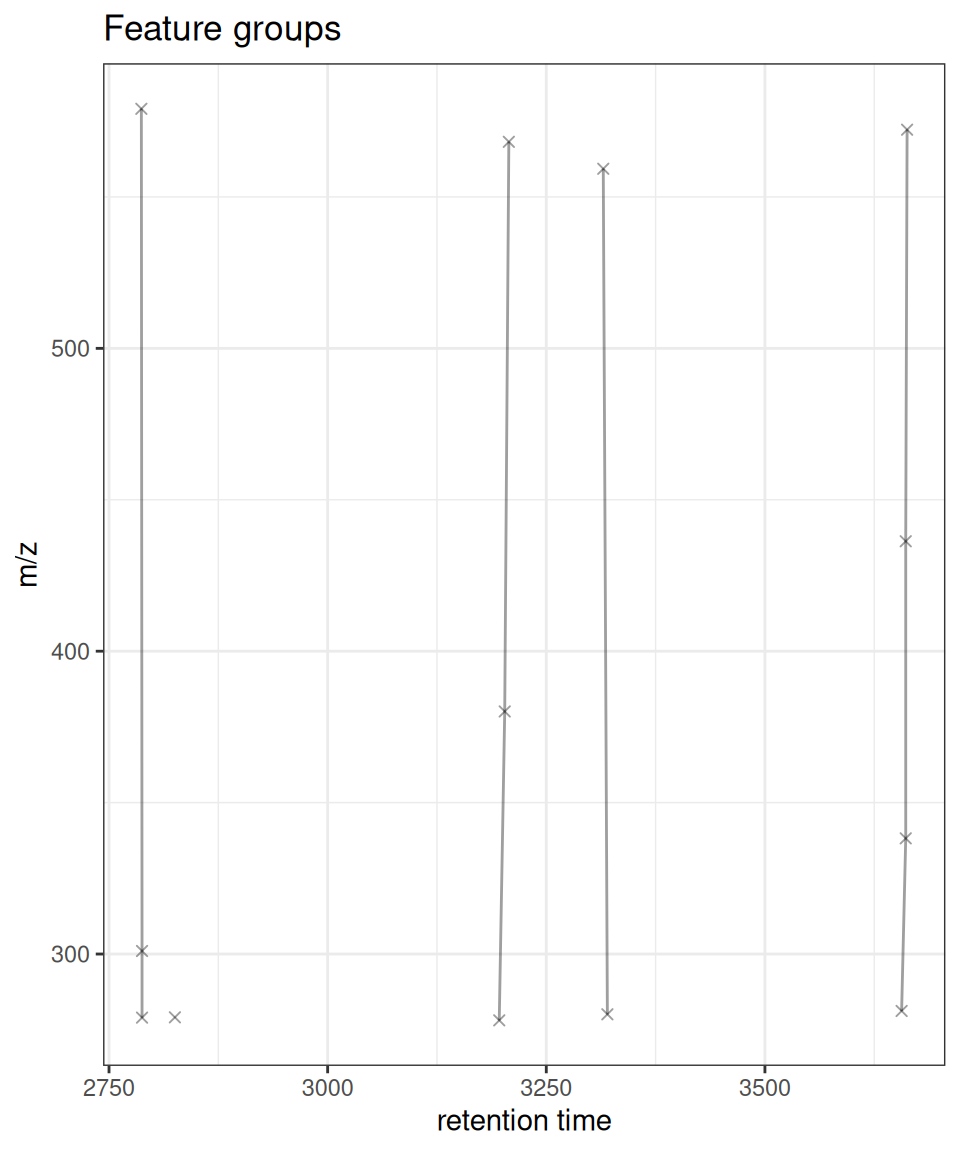
API Differences
Unlike the original xcms
plotFeatureGroups(), the ggplot2 version does not havexlab,ylab, ormainparameters. Instead, use ggplot2’slabs()function to customize labels after plot creation:# Customize labels with labs() fg_subset <- unique(featureGroups(xdata))[1:2] gplotFeatureGroups(xdata, featureGroups = fg_subset) + labs(x = "Retention Time (s)", y = "Mass/Charge", title = "My Custom Title")This follows ggplot2 conventions and makes the API more consistent with the broader ggplot2 ecosystem.
Session Info
sessionInfo()
#> R version 4.6.0 (2026-04-24)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] plotly_4.12.0 patchwork_1.3.2 ggplot2_4.0.3
#> [4] MsFeatures_1.20.0 MsExperiment_1.14.0 ProtGenerics_1.44.0
#> [7] xcmsVis_0.99.11 xcms_4.10.0 BiocParallel_1.46.0
#>
#> loaded via a namespace (and not attached):
#> [1] DBI_1.3.0 rlang_1.2.0
#> [3] magrittr_2.0.5 clue_0.3-68
#> [5] MassSpecWavelet_1.78.0 otel_0.2.0
#> [7] matrixStats_1.5.0 compiler_4.6.0
#> [9] PTMods_1.0.0 vctrs_0.7.3
#> [11] reshape2_1.4.5 stringr_1.6.0
#> [13] pkgconfig_2.0.3 MetaboCoreUtils_1.20.1
#> [15] crayon_1.5.3 fastmap_1.2.0
#> [17] XVector_0.52.0 labeling_0.4.3
#> [19] rmarkdown_2.31 preprocessCore_1.74.0
#> [21] purrr_1.2.2 xfun_0.57
#> [23] MultiAssayExperiment_1.38.0 jsonlite_2.0.0
#> [25] progress_1.2.3 DelayedArray_0.38.1
#> [27] parallel_4.6.0 prettyunits_1.2.0
#> [29] cluster_2.1.8.2 R6_2.6.1
#> [31] stringi_1.8.7 RColorBrewer_1.1-3
#> [33] limma_3.68.1 GenomicRanges_1.64.0
#> [35] Rcpp_1.1.1-1.1 Seqinfo_1.2.0
#> [37] SummarizedExperiment_1.42.0 iterators_1.0.14
#> [39] knitr_1.51 IRanges_2.46.0
#> [41] Matrix_1.7-5 igraph_2.3.1
#> [43] tidyselect_1.2.1 abind_1.4-8
#> [45] yaml_2.3.12 doParallel_1.0.17
#> [47] codetools_0.2-20 affy_1.90.0
#> [49] lattice_0.22-9 tibble_3.3.1
#> [51] plyr_1.8.9 withr_3.0.2
#> [53] Biobase_2.72.0 S7_0.2.2
#> [55] evaluate_1.0.5 Spectra_1.22.0
#> [57] pillar_1.11.1 affyio_1.82.0
#> [59] BiocManager_1.30.27 MatrixGenerics_1.24.0
#> [61] foreach_1.5.2 stats4_4.6.0
#> [63] MSnbase_2.37.0 MALDIquant_1.22.3
#> [65] ncdf4_1.24 generics_0.1.4
#> [67] S4Vectors_0.50.0 hms_1.1.4
#> [69] scales_1.4.0 glue_1.8.1
#> [71] lazyeval_0.2.3 tools_4.6.0
#> [73] mzID_1.50.0 data.table_1.18.2.1
#> [75] QFeatures_1.22.0 vsn_3.80.0
#> [77] mzR_2.46.0 fs_2.1.0
#> [79] XML_3.99-0.23 grid_4.6.0
#> [81] impute_1.86.0 tidyr_1.3.2
#> [83] crosstalk_1.2.2 MsCoreUtils_1.24.0
#> [85] PSMatch_1.16.0 cli_3.6.6
#> [87] viridisLite_0.4.3 S4Arrays_1.12.0
#> [89] dplyr_1.2.1 AnnotationFilter_1.36.0
#> [91] pcaMethods_2.4.0 gtable_0.3.6
#> [93] digest_0.6.39 BiocGenerics_0.58.0
#> [95] SparseArray_1.12.2 htmlwidgets_1.6.4
#> [97] farver_2.1.2 htmltools_0.5.9
#> [99] lifecycle_1.0.5 httr_1.4.8
#> [101] statmod_1.5.1 MASS_7.3-65